Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Charles Tiu | -- | 1669 | 2022-06-05 12:35:58 | | | |
| 2 | Sirius Huang | -1 word(s) | 1668 | 2022-06-06 04:56:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tiu, C.; Zhu, F.; Wang, L.; De Alwis, R. Phage ImmunoPrecipitation Sequencing (PhIP-Seq). Encyclopedia. Available online: https://encyclopedia.pub/entry/23729 (accessed on 26 July 2026).
Tiu C, Zhu F, Wang L, De Alwis R. Phage ImmunoPrecipitation Sequencing (PhIP-Seq). Encyclopedia. Available at: https://encyclopedia.pub/entry/23729. Accessed July 26, 2026.
Tiu, Charles, Feng Zhu, Lin-Fa Wang, Ruklanthi De Alwis. "Phage ImmunoPrecipitation Sequencing (PhIP-Seq)" Encyclopedia, https://encyclopedia.pub/entry/23729 (accessed July 26, 2026).
Tiu, C., Zhu, F., Wang, L., & De Alwis, R. (2022, June 05). Phage ImmunoPrecipitation Sequencing (PhIP-Seq). In Encyclopedia. https://encyclopedia.pub/entry/23729
Tiu, Charles, et al. "Phage ImmunoPrecipitation Sequencing (PhIP-Seq)." Encyclopedia. Web. 05 June, 2022.
Copy Citation
Phage ImmunoPrecipitation Sequencing (PhIP-Seq) is a serological technology that is revolutionizing the manner in which we track antibody profiles. It is an antigen-specific antibody detection assay that is high throughput, highly sensitive, and mega-plexable (i.e., ability of plexing over a large number of different peptides).
phage-immunoprecipitation sequencing
PhIP-Seq
phage
antibody
Phage ImmunoPrecipitation Sequencing (PhIP-Seq) is an antigen-specific antibody detection assay that is high throughput, highly sensitive, and mega-plexable (i.e., ability of plexing over a large number of different peptides). Numerous PhIP-Seq libraries panning different types of proteins have been developed. Following the seminal study identifying autoantigens [1], PhIP-Seq has since been expanded to include infectious diseases, such as viruses (including numerous human viruses, arthropod-borne viruses, SARS-CoV-2, etc.) [2][3][4], and bacteria (mostly in the form of human microbiota) [5]. A schematic representation of the typical PhIP-Seq workflow is displayed in Figure 1 [6].
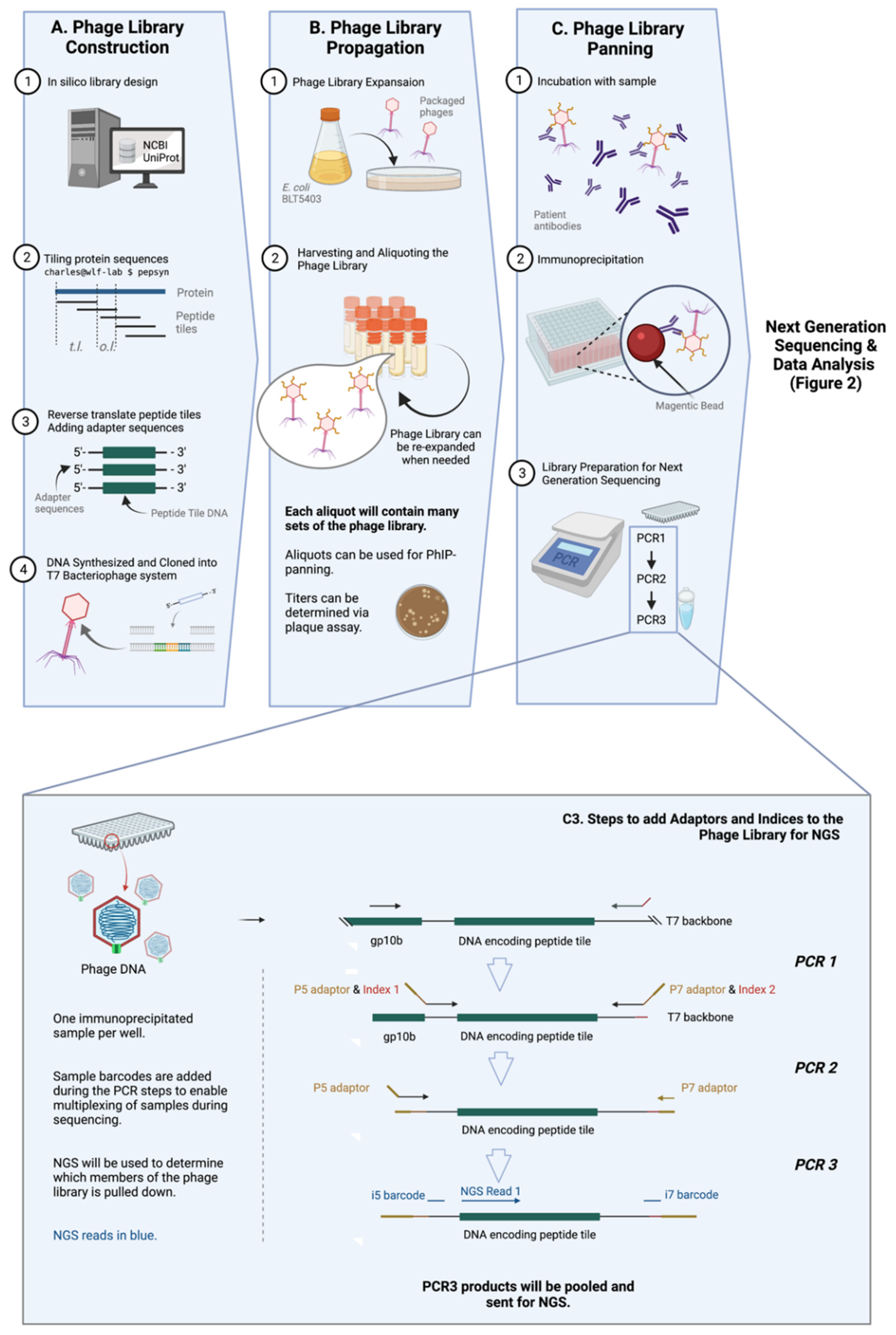
Figure 1. General workflow of PhIP-Seq. The PhIP-Seq methodology is composed of four key steps: (A) phage library construction, (B) phage library propagation, (C) phage library panning, and data analysis (displayed in Figure 2). Addition of adapters and indices to immunoprecipitated phage sample using PCR (step C3 of Figure 1A is described in detail). Abbreviations: tile length (t.l.), overlap length (o.l.). Created with BioRender.com.
1. Phage Library Construction
Library design is rational [5] and does not require tissue, transcripts, directional cloning, or in-frame open reading frames (ORFs) determination. Almost any genetic sequence, regardless of its identity and whether it has been previously expressed or purified, can be cloned and tested via PhIP-Seq. This versatility allows for numerous known strains and infectious pathogens (viruses [7][8], and possibly parasites and fungi, etc.), as well as relatively new pathogens to be easily included in PhIP-Seq libraries when available. Libraries containing targets to novel pathogens, like SARS-CoV-2, can be made with relative ease, as seen in Shrock et al. [2] (Figure 1A).
One of the key improvements of PhIP-Seq is its use of an in-silico designed and custom-built library. Unlike traditional phage display, PhIP-Seq’s library is composed of sequences with defined lengths, overlaps, and a known annotation (e.g., 56 amino acid tiles with 28 amino acid overlap, as in the case for the VirScan—one of the earliest PhIP-Seq libraries) [3]. The tools (pepsyn, a Python-based tool that designs peptide libraries) and the shell codes required for the design of these customized libraries are readily available online [6]. Briefly, the set of codes converts the input amino acid sequences (in the fasta file format) to nucleic acid sequences that codes for the corresponding peptide set tile length and tile overlap. The sequences can then be synthesized using a microarray-based DNA synthesis platform, which may then be cloned into a commercially-available T7 phage display system.
2. Phage Library Propagation
The PhIP-Seq system utilizes the T7 bacteriophage, a lytic phage, as its powerhouse to express antigens for detection. T7 bacteriophage expresses copies of the target epitope or peptide on its surface, and the identity of this target epitope is reflected in its genomic sequence. PhIP-Seq is composed of ‘live’ phages, i.e., replication-competent phages [9]. Phages (Figure 1B) are expanded within a specified bacterial host grown on solid phase, and progeny phages in supernatant are collected. These progeny phages will carry the same genetic information and therefore, the same corresponding peptide tiles on their surface as the parental phages. Upon expansion, the phage library can be used for further expansion or for PhIP-Seq experiments.
3. Phage Immunoprecipitation
Immunoprecipitation of PhIP-Seq phages with antibodies is typically straightforward and not very different from traditional phage display. Patient or animal [10] sera or cerebrospinal fluid (CSF) (or theoretically, any biological fluid containing antibodies, such as saliva, urine, etc.) can be used to probe PhIP-Seq libraries. Biological samples are typically first measured by ELISA to ensure the presence of antigen-specific immunoglobulin. This is followed by the incubation of antibody-containing sample and the peptide-displaying phages, and then an immunoprecipitation step which typically involves protein A/G on magnetic beads. (Figure 1C) More detailed investigations into isotype- (i.e., IgG, IgA, IgM or IgE, etc.) or subclass (i.e., IgG1, IgG2, IgG3, IgG4 etc.)-specific humoral responses can also be conducted by pulling down using beads coated with the relevant anti-isotype or anti-subclass monoclonal antibody [11] or reagents. For example, in one published study, the authors used streptavidin-coupled magnetic beads with biotin-conjugated omalizumab to pull down and study IgE-specific responses [12].
Before the identity of immunoprecipitated phages can then be determined with high throughput sequencing, a few steps of pre-processing are required. This involves amplifying the peptide tile specific sequence of the phage genome by polymerase chain reaction (PCR), subsequently adding sample barcodes and eventually the next generation sequencing (NGS) adaptors, e.g., P5 and P7 (Figure 1C and the inset). Sample barcodes used to identify individual samples as PhIP-Seq runs are typically multiplexed given the read depths of modern NGS systems.
The high sensitivity provided by using molecular sequencing as the detection method, can lead to some assay background. To account for background signal, each run is typically supplemented with a number of negative controls (i.e., immunoprecipitation of phage library in the presence of PBS only) and library controls (i.e., where only the input library is sequenced). The former captures the background due to direct phage-bead interaction in the absence of antibodies or components from biological samples, while the latter captures the available breadth of the library. Additionally, technical duplicates can also be run for each sample and averaged to improve accuracy [7][13]. In other works, antibody binding to rare or uncommon viruses (such as Rabies and Ebola virus) are used as baseline controls for samples to account for sample-to-sample variations in sequencing depth [14]. Intra-subject comparison of time-course samples (i.e., samples taken from pre- and post-infection) can also be used to account for baseline background signal and can more clearly show the changes in the antibody repertoire brought upon by a specific infection or exposure event [15].
4. Data Analysis Overview
There is no unified pipeline or script for the analysis of PhIP-Seq data. However, several research groups are trying to establish computational pipelines and make it available online for general use (https://github.com/lasersonlab/phip-stat, accessed on 20 April 2022), (phippery, https://github.com/matsengrp/phippery and phip-flow, https://github.com/matsengrp/phip-flow, both accessed on 20 April 2022).
Figure 2 illustrates a typical pipeline of data analysis; here is an example used by the pipeline, phip-stat (https://github.com/lasersonlab/phip-stat, accessed on 8 May 2022). High throughput sequencing data is demultiplexed to identify PhIP-Seq data specific to each sample and aligned to reference sequences generated from the original sequence files to deconvolute the peptide IDs. The number of reads for each specific peptide tile is then counted for each sample, which is followed by normalization with the read (either to a set number of reads per sample or using a mathematical model). Typically, normalized read counts will be reported and this can then be used for downstream analysis. Following normalization, some studies will apply additional statistical testing based on poison distribution (and use a p-value-based metric (−log10(p)) [3][8][16], or utilize a z-score metric to describe enrichment of peptide tiles [14][15][17]). The resulting metrics may then be used to analyze and visualize the phage immunoprecipitation results.
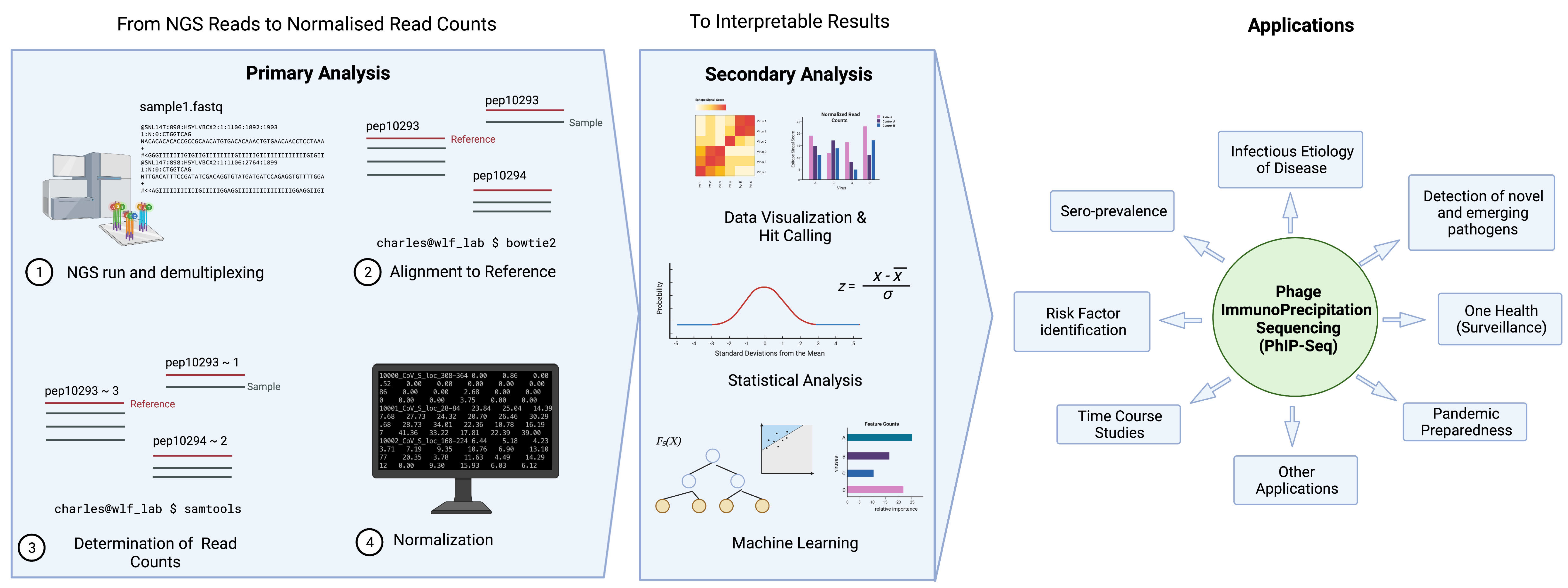
Figure 2. General data analysis pipeline and applications of PhIP-Seq. Created with BioRender.com.
There are two key steps involved in converting NGS reads to interpretable results. Primary analysis entails alignment of reads to reference sequences, assessment of read counts, and data normalization. Depending on the scientific question and application, data will undergo secondary analysis in the form of data visualization, statistical analysis, and machine learning, etc.
5. Determining the Hits
Calculating a z-score metric or a p-value metric may not be sufficiently informative as investigators are typically more interested in the actual significance of the result—i.e., if it indicates a prior exposure or otherwise.
A key problem with the use of high throughput serology is not having a “gold standard comparator”—many of the target proteins typically will not have commercial serological tests available. Without a standard comparator to allow us to “train” the platform, it will be challenging to determine which z-score, or p-value would correspond a “true hit” or a prior exposure. Thus, researchers frequently use statistical measures and controls to determine which peptides or viruses are considered positive for a PhIP-Seq run. Cut-offs are sometimes defined, for example, based on a reproducibility threshold based on the −log10(p-value) of technical duplicates, and an epitope is considered to be positive when this threshold is crossed [16]. Confidence to the scoring algorithm and cut-offs is typically bolstered with the measures of antibody response to common human infections (e.g., rhinovirus, or Epstein-Barr virus infections), cross-comparison against conventional serological assays, or provided for by an infection history (for example, clinically diagnosed infected patients). Statistical tests such as t-tests [7] and the Mann–Whitney test [4][8][16][18], amongst others, are also frequently used to compare data generated from PhIP-Seq.
6. Other Analysis Strategies (Machine Learning, AVARDA, Novel Pipelines)
More advanced statistical tools are sometimes used to analyze and interpret results from PhIP-Seq. In some studies, authors use gradient boosting algorithm xgboost [19] to determine important peptide tiles (or features) that distinguish one group of patients from another [2][5][16]. This specially works well for case-control studies with large well-defined study populations.
Another involves the use of epitope similarity across different peptide tiles. Monaco et al. attempted to improve the VirScan platform by accounting for possible cross-reactivity among peptide tiles by sequence alignment. This technique, named Antiviral Antibody Response Deconvolution Algorithm (AVARDA), is based on the premise that antibodies can cross-react with similar peptide tiles assigned to different viruses. Antibody cross-reactivity between viruses has not been accounted for in prior PhIP-Seq analysis strategies, and this valuable information is usually lost during analysis [20].
7. Programming Language and Skills Needed
Primary and secondary data analysis require basic knowledge of command line interface and access to a decent computing infrastructure [6]. Most data analysis can be conducted with Python or R scripts. Having a working knowledge on these programming languages, or having access to individuals who are experienced in data science or bioinformatics, will be of great utility to investigators utilizing PhIP-Seq as a serological tool.
References
- Larman, H.B.; Zhao, Z.; Laserson, U.; Li, M.Z.; Ciccia, A.; Gakidis, M.A.; Church, G.M.; Kesari, S.; Leproust, E.M.; Solimini, N.L.; et al. Autoantigen discovery with a synthetic human peptidome. Nat. Biotechnol. 2011, 29, 535–541.
- Shrock, E.; Fujimura, E.; Kula, T.; Timms, R.T.; Lee, I.H.; Leng, Y.; Robinson, M.L.; Sie, B.M.; Li, M.Z.; Chen, Y.; et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science 2020, 370, eabd4250.
- Xu, G.J.; Kula, T.; Xu, Q.; Li, M.Z.; Vernon, S.D.; Ndung’u, T.; Ruxrungtham, K.; Sanchez, J.; Brander, C.; Chung, R.T.; et al. Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science 2015, 348, aaa0698.
- Schubert, R.D.; Hawes, I.A.; Ramachandran, P.S.; Ramesh, A.; Crawford, E.D.; Pak, J.E.; Wu, W.; Cheung, C.K.; O’Donovan, B.D.; Tato, C.M.; et al. Pan-viral serology implicates enteroviruses in acute flaccid myelitis. Nat. Med. 2019, 25, 1748–1752.
- Vogl, T.; Klompus, S.; Leviatan, S.; Kalka, I.N.; Weinberger, A.; Wijmenga, C.; Fu, J.; Zhernakova, A.; Weersma, R.K.; Segal, E. Population-wide diversity and stability of serum antibody epitope repertoires against human microbiota. Nat. Med. 2021, 27, 1442–1450.
- Mohan, D.; Wansley, D.L.; Sie, B.M.; Noon, M.S.; Baer, A.N.; Laserson, U.; Larman, H.B. PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat. Protoc. 2018, 13, 1958–1978.
- Mina, M.J.; Kula, T.; Leng, Y.; Li, M.; de Vries, R.D.; Knip, M.; Siljander, H.; Rewers, M.; Choy, D.F.; Wilson, M.S.; et al. Measles virus infection diminishes preexisting antibodies that offer protection from other pathogens. Science 2019, 366, 599–606.
- Hasan, M.R.; Rahman, M.; Khan, T.; Saeed, A.; Sundararaju, S.; Flores, A.; Hawken, P.; Rawat, A.; Elkum, N.; Hussain, K.; et al. Virome-wide serological profiling reveals association of herpesviruses with obesity. Sci. Rep. 2021, 11, 2562.
- Li, W.; Caberoy, N.B. New perspective for phage display as an efficient and versatile technology of functional proteomics. Appl. Microbiol. Biotechnol. 2010, 85, 909–919.
- Irving, A.T.; Rozario, P.; Kong, P.-S.; Luko, K.; Gorman, J.J.; Hastie, M.L.; Chia, W.N.; Mani, S.; Lee, B.P.Y.H.; Smith, G.J.D.; et al. Robust dengue virus infection in bat cells and limited innate immune responses coupled with positive serology from bats in IndoMalaya and Australasia. Cell. Mol. Life Sci. 2020, 77, 1607–1622.
- Ravichandran, S.; Hahn, M.; Belaunzarán-Zamudio, P.F.; Ramos-Castañeda, J.; Nájera-Cancino, G.; Caballero-Sosa, S.; Navarro-Fuentes, K.R.; Ruiz-Palacios, G.; Golding, H.; Beigel, J.H.; et al. Differential human antibody repertoires following Zika infection and the implications for serodiagnostics and disease outcome. Nat. Commun. 2019, 10, 1943.
- Monaco, D.R.; Sie, B.M.; Nirschl, T.R.; Knight, A.C.; Sampson, H.A.; Nowak-Wegrzyn, A.; Wood, R.A.; Hamilton, R.G.; Frischmeyer-Guerrerio, P.A.; Larman, H.B. Profiling serum antibodies with a pan allergen phage library identifies key wheat allergy epitopes. Nat. Commun. 2021, 12, 379.
- Stoddard, C.I.; Galloway, J.; Chu, H.Y.; Shipley, M.M.; Sung, K.; Itell, H.L.; Wolf, C.R.; Logue, J.K.; Magedson, A.; Garrett, M.E.; et al. Epitope profiling reveals binding signatures of SARS-CoV-2 immune response in natural infection and cross-reactivity with endemic human CoVs. Cell Rep. 2021, 35, 109164.
- Eshleman, S.H.; Laeyendecker, O.; Kammers, K.; Chen, A.; Sivay, M.V.; Kottapalli, S.; Sie, B.M.; Yuan, T.; Monaco, D.R.; Mohan, D.; et al. Comprehensive Profiling of HIV Antibody Evolution. Cell Rep. 2019, 27, 1422–1433.e4.
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301.
- Liu, J.; Tang, W.; Budhu, A.; Forgues, M.; Hernandez, M.O.; Candia, J.; Kim, Y.; Bowman, E.D.; Ambs, S.; Zhao, Y.; et al. A Viral Exposure Signature Defines Early Onset of Hepatocellular Carcinoma. Cell 2020, 182, 317–328.e10.
- Johnson, T.P.; Larman, H.B.; Lee, M.H.; Whitehead, S.S.; Kowalak, J.; Toro, C.; Lau, C.C.; Kim, J.; Johnson, K.R.; Reoma, L.B.; et al. Chronic Dengue Virus Panencephalitis in a Patient with Progressive Dementia with Extrapyramidal Features. Ann. Neurol. 2019, 86, 695–703.
- Leon, K.E.; Schubert, R.D.; Casas-Alba, D.; Hawes, I.A.; Ramachandran, P.S.; Ramesh, A.; Pak, J.E.; Wu, W.; Cheung, C.K.; Crawford, E.D.; et al. Genomic and serologic characterization of enterovirus A71 brainstem encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e703.
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794.
- Monaco, D.R.; Kottapalli, S.V.; Breitwieser, F.P.; Anderson, D.E.; Wijaya, L.; Tan, K.; Chia, W.N.; Kammers, K.; Caturegli, P.; Waugh, K.; et al. Deconvoluting virome-wide antibody epitope reactivity profiles. eBioMedicine 2021, 75, 103747.
More
Information
Subjects:
Infectious Diseases; Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.2K
Revisions:
2 times
(View History)
Update Date:
06 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No