 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Thirumala-Devi Kanneganti | -- | 2851 | 2022-06-02 02:56:46 | | | |
| 2 | Camila Xu | + 125 word(s) | 2976 | 2022-06-02 04:51:55 | | |
Video Upload Options
Consideration of the totality of biological effects from cell death in multiple studies has led to the conceptualization of PANoptosis, a unique inflammatory cell death pathway that integrates components from other cell death pathways. PANoptosis is implicated in driving innate immune responses and inflammation and cannot be individually accounted for by pyroptosis, apoptosis, or necroptosis alone. PANoptosis is regulated by multifaceted macromolecular complexes called PANoptosomes.
1. Programmed Cell Death and PANoptosis
Key Components in Inflammatory Programmed Cell Death Pathways
2. Historical Development of the PANoptosis Concept
3. Prototypical Examples of PANoptosis
Here, researchers focus on the most mechanistically well-characterized examples of PANoptosis and PANoptosomes [3][7][9][10][11][12][20] (Figure 1).
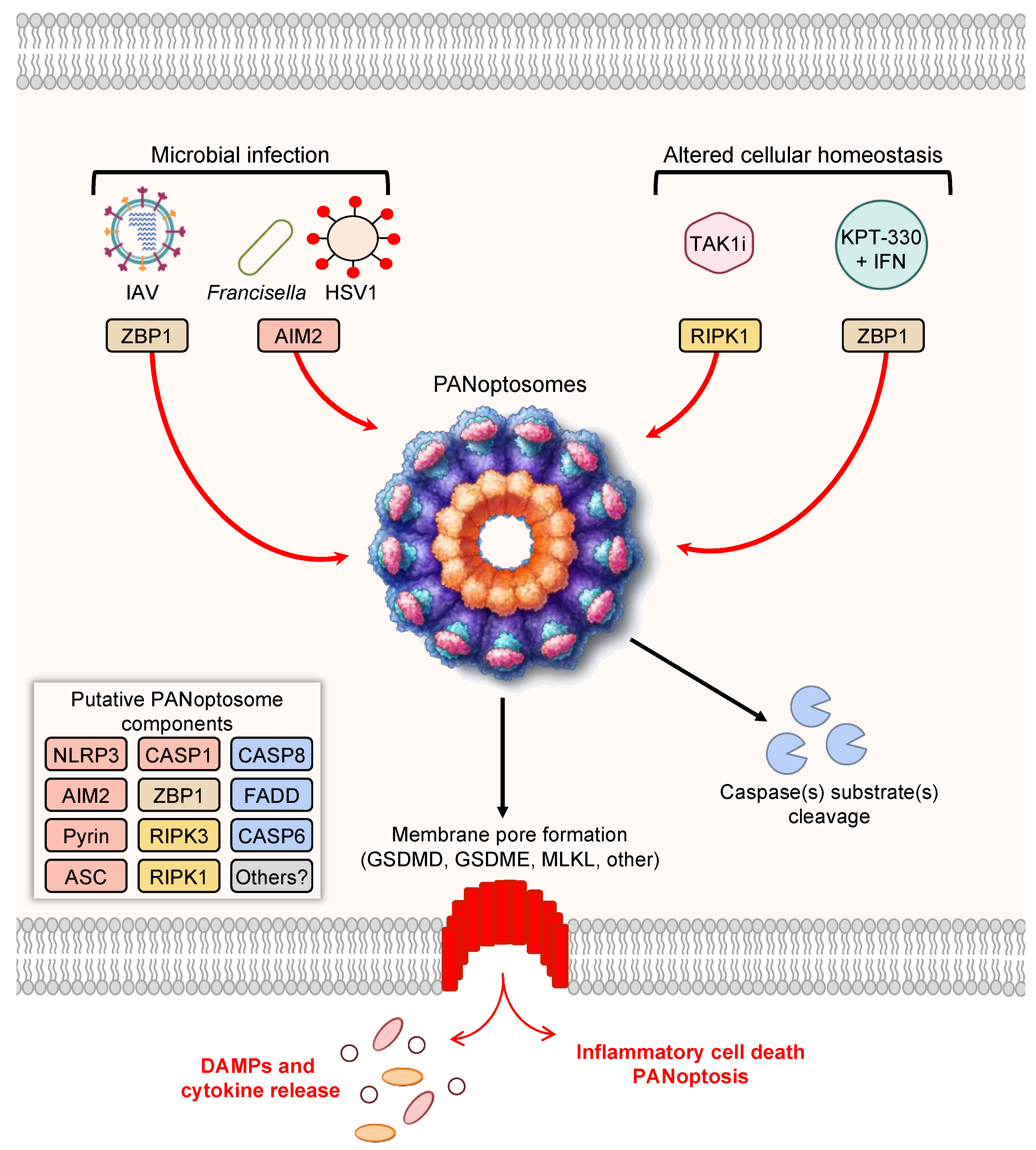
Figure 1. PANoptosis and PANoptosome formation. Upon exposure to cellular insults, such as microbial infection or altered cellular homeostasis, sensors can detect the perturbation and activate PANoptosis. Prototypical examples of PANoptosis are depicted here. Sensor activation can lead to the formation of a multiprotein complex, the PANoptosome. PANoptosomes have the potential to bring together diverse components from previously segregated cell death pathways. These may be dynamic complexes, and their protein composition may vary in trigger- and time-dependent manners. Potential PANoptosome components putatively include inflammasome sensors, such as nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3), absent in melanoma 2 (AIM2), Pyrin, Z-DNA-binding protein 1 (ZBP1), or others; apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC); caspase-1 (CASP1); receptor-interacting serine/threonine protein kinase 3 (RIPK3); RIPK1; caspase-8 (CASP8); Fas-associated protein with death domain (FADD); and/or caspase-6 (CASP6). PANoptosis involves membrane pore formation for the execution of cell death to release cytokines, such as IL-1β and IL-18, and DAMPs. Figure created with https://biorender.com/ (accessed on 17 March 2022).
4. PANoptosis Regulation via IRF1
5. Implications of PANoptosis in Disease
PANoptosis has been implicated across the disease spectrum, including in cerebral ischemia [114]; bacterial, viral, and fungal infections [3][9][10][11][12][13][14][15], including oral infections [115][116]; inflammatory diseases [2][5][16][18][20]; cancers [4][7][8]; and cancer therapies [4][7][117][118]. It will be important to improve the understanding of this pathway and identify how previous descriptions of crosstalk, plasticity, redundancies, molecular switches, and interconnectedness among cell death processes fit within this inclusive concept to gain a holistic picture of cell death. Only when researchers identify all the ingredients in the PAN can researchers begin to effectively target these molecules and develop novel therapeutics to save lives and improve patient outcomes.
References
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 2019, 9, 406.
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e117.
- Malireddi, R.K.S.; Kesavardhana, S.; Karki, R.; Kancharana, B.; Burton, A.R.; Kanneganti, T.D. RIPK1 Distinctly Regulates Yersinia-Induced Inflammatory Cell Death, PANoptosis. Immunohorizons 2020, 4, 789–796.
- Malireddi, R.K.S.; Karki, R.; Sundaram, B.; Kancharana, B.; Lee, S.; Samir, P.; Kanneganti, T.D. Inflammatory Cell Death, PANoptosis, Mediated by Cytokines in Diverse Cancer Lineages Inhibits Tumor Growth. Immunohorizons 2021, 5, 568–580.
- Malireddi, R.K.S.; Gurung, P.; Mavuluri, J.; Dasari, T.K.; Klco, J.M.; Chi, H.; Kanneganti, T.D. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 2018, 215, 1023–1034.
- Malireddi, R.K.; Ippagunta, S.; Lamkanfi, M.; Kanneganti, T.D. Cutting edge: Proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J. Immunol. 2010, 185, 3127–3130.
- Karki, R.; Sundaram, B.; Sharma, B.R.; Lee, S.; Malireddi, R.K.S.; Nguyen, L.N.; Christgen, S.; Zheng, M.; Wang, Y.; Samir, P.; et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 2021, 37, 109858.
- Karki, R.; Sharma, B.R.; Lee, E.; Banoth, B.; Malireddi, R.K.S.; Samir, P.; Tuladhar, S.; Mummareddy, H.; Burton, A.R.; Vogel, P.; et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI Insight 2020, 5, e136720.
- Kuriakose, T.; Man, S.M.; Subbarao Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045.
- Christgen, S.; Zheng, M.; Kesavardhana, S.; Karki, R.; Malireddi, R.K.S.; Banoth, B.; Place, D.E.; Briard, B.; Sharma, B.R.; Tuladhar, S.; et al. Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020, 10, 237.
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.D. Caspase-6 is a key regulator of innate immunity, inflammasome activation and host defense. Cell 2020, 181, 674–687.e13.
- Lee, S.; Karki, R.; Wang, Y.; Nguyen, L.N.; Kalathur, R.C.; Kanneganti, T.D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 2021, 597, 415–419.
- Kesavardhana, S.; Malireddi, R.K.S.; Burton, A.R.; Porter, S.N.; Vogel, P.; Pruett-Miller, S.M.; Kanneganti, T.D. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J. Biol. Chem. 2020, 295, 8325–8330.
- Banoth, B.; Tuladhar, S.; Karki, R.; Sharma, B.R.; Briard, B.; Kesavardhana, S.; Burton, A.; Kanneganti, T.D. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J. Biol. Chem. 2020, 295, 18276–18283.
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052.
- Lukens, J.R.; Gurung, P.; Vogel, P.; Johnson, G.R.; Carter, R.A.; McGoldrick, D.J.; Bandi, S.R.; Calabrese, C.R.; Walle, L.V.; Lamkanfi, M.; et al. Dietary modulation of the microbiome affects autoinflammatory disease. Nature 2014, 516, 246–249.
- Lamkanfi, M.; Kanneganti, T.D.; Van Damme, P.; Vanden Berghe, T.; Vanoverberghe, I.; Vandekerckhove, J.; Vandenabeele, P.; Gevaert, K.; Nunez, G. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell. Proteom. 2008, 7, 2350–2363.
- Gurung, P.; Burton, A.; Kanneganti, T.D. NLRP3 inflammasome plays a redundant role with caspase 8 to promote IL-1β–mediated osteomyelitis. Proc. Natl. Acad. Sci. USA 2016, 113, 4452–4457.
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846.
- Malireddi, R.K.S.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H.; Vogel, P.; Pelletier, S.; Burgula, S.; Kanneganti, T.D. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020, 217, e20191644.
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114.
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426.
- Kanneganti, T.D.; Ozoren, N.; Body-Malapel, M.; Amer, A.; Park, J.H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006, 440, 233–236.
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232.
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241.
- Franchi, L.; Amer, A.; Body-Malapel, M.; Kanneganti, T.D.; Ozoren, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 2006, 7, 576–582.
- Miao, E.A.; Alpuche-Aranda, C.M.; Dors, M.; Clark, A.E.; Bader, M.W.; Miller, S.I.; Aderem, A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 2006, 7, 569–575.
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218.
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241.
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513.
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518.
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.X.; Halfmann, R.; Chen, Z.J. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014, 156, 1207–1222.
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206.
- Masumoto, J.; Taniguchi, S.; Ayukawa, K.; Sarvotham, H.; Kishino, T.; Niikawa, N.; Hidaka, E.; Katsuyama, T.; Higuchi, T.; Sagara, J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 1999, 274, 33835–33838.
- Sborgi, L.; Ravotti, F.; Dandey, V.P.; Dick, M.S.; Mazur, A.; Reckel, S.; Chami, M.; Scherer, S.; Huber, M.; Böckmann, A.; et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 13237–13242.
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298.
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863.
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116.
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158.
- Sborgi, L.; Ruhl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671.
- Kayagaki, N.; Lee, B.L.; Stowe, I.B.; Kornfeld, O.S.; O’Rourke, K.; Mirrashidi, K.M.; Haley, B.; Watanabe, C.; Roose-Girma, M.; Modrusan, Z.; et al. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci. Signal. 2019, 12, eaax4917.
- Benaoudia, S.; Martin, A.; Puig Gamez, M.; Gay, G.; Lagrange, B.; Cornut, M.; Krasnykov, K.; Claude, J.B.; Bourgeois, C.F.; Hughes, S.; et al. A genome-wide screen identifies IRF2 as a key regulator of caspase-4 in human cells. EMBO Rep. 2019, 20, e48235.
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257.
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413.
- Kim, H.E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550.
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489.
- Boldin, M.P.; Goncharov, T.M.; Goltsev, Y.V.; Wallach, D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996, 85, 803–815.
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O’Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell 1996, 85, 817–827.
- Gross, A.; Yin, X.M.; Wang, K.; Wei, M.C.; Jockel, J.; Milliman, C.; Erdjument-Bromage, H.; Tempst, P.; Korsmeyer, S.J. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J. Biol. Chem. 1999, 274, 1156–1163.
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490.
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501.
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000, 14, 2060–2071.
- Kuwana, T.; Mackey, M.R.; Perkins, G.; Ellisman, M.H.; Latterich, M.; Schneiter, R.; Green, D.R.; Newmeyer, D.D. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002, 111, 331–342.
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol Cancer 2019, 18, 100.
- Nailwal, H.; Chan, F.K. Necroptosis in anti-viral inflammation. Cell Death Differ. 2019, 26, 4–13.
- Kang, T.B.; Ben-Moshe, T.; Varfolomeev, E.E.; Pewzner-Jung, Y.; Yogev, N.; Jurewicz, A.; Waisman, A.; Brenner, O.; Haffner, R.; Gustafsson, E.; et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol. 2004, 173, 2976–2984.
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431.
- O’Donnell, M.A.; Perez-Jimenez, E.; Oberst, A.; Ng, A.; Massoumi, R.; Xavier, R.; Green, D.R.; Ting, A.T. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 2011, 13, 1437–1442.
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007, 19, 2056–2067.
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195.
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714.
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227.
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327.
- Garcia, L.R.; Tenev, T.; Newman, R.; Haich, R.O.; Liccardi, G.; John, S.W.; Annibaldi, A.; Yu, L.; Pardo, M.; Young, S.N.; et al. Ubiquitylation of MLKL at lysine 219 positively regulates necroptosis-induced tissue injury and pathogen clearance. Nat. Commun. 2021, 12, 3364.
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343.
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258.
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J. Biol. Chem. 2016, 291, 103–109.
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357.
- Samir, P.; Kesavardhana, S.; Patmore, D.M.; Gingras, S.; Malireddi, R.K.S.; Karki, R.; Guy, C.S.; Briard, B.; Place, D.E.; Bhattacharya, A.; et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 2019, 573, 590–594.
- Kuriakose, T.; Zheng, M.; Neale, G.; Kanneganti, T.D. IRF1 Is a Transcriptional Regulator of ZBP1 Promoting NLRP3 Inflammasome Activation and Cell Death during Influenza Virus Infection. J. Immunol. 2018, 200, 1489–1495.
- Karki, R.; Lee, E.; Place, D.; Samir, P.; Mavuluri, J.; Sharma, B.R.; Balakrishnan, A.; Malireddi, R.K.S.; Geiger, R.; Zhu, Q.; et al. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 2018, 173, 920–933.e13.
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136.
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327.
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol. Immunol. 2021, 18, 1106–1121.
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495.
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367.
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372.
- Alvarez-Diaz, S.; Dillon, C.P.; Lalaoui, N.; Tanzer, M.C.; Rodriguez, D.A.; Lin, A.; Lebois, M.; Hakem, R.; Josefsson, E.C.; O’Reilly, L.A.; et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513–526.
- Varfolomeev, E.E.; Schuchmann, M.; Luria, V.; Chiannilkulchai, N.; Beckmann, J.S.; Mett, I.L.; Rebrikov, D.; Brodianski, V.M.; Kemper, O.C.; Kollet, O.; et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998, 9, 267–276.
- Lalaoui, N.; Boyden, S.E.; Oda, H.; Wood, G.M.; Stone, D.L.; Chau, D.; Liu, L.; Stoffels, M.; Kratina, T.; Lawlor, K.E.; et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 2020, 577, 103–108.
- Chun, H.J.; Zheng, L.; Ahmad, M.; Wang, J.; Speirs, C.K.; Siegel, R.M.; Dale, J.K.; Puck, J.; Davis, J.; Hall, C.G.; et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 2002, 419, 395–399.
- Puri, A.W.; Broz, P.; Shen, A.; Monack, D.M.; Bogyo, M. Caspase-1 activity is required to bypass macrophage apoptosis upon Salmonella infection. Nat. Chem. Biol. 2012, 8, 745–747.
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519.
- Pierini, R.; Juruj, C.; Perret, M.; Jones, C.L.; Mangeot, P.; Weiss, D.S.; Henry, T. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012, 19, 1709–1721.
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160.
- Man, S.M.; Tourlomousis, P.; Hopkins, L.; Monie, T.P.; Fitzgerald, K.A.; Bryant, C.E. Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1β production. J. Immunol. 2013, 191, 5239–5246.
- Mascarenhas, D.P.A.; Cerqueira, D.M.; Pereira, M.S.F.; Castanheira, F.V.S.; Fernandes, T.D.; Manin, G.Z.; Cunha, L.D.; Zamboni, D.S. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog. 2017, 13, e1006502.
- Van Opdenbosch, N.; Van Gorp, H.; Verdonckt, M.; Saavedra, P.H.V.; de Vasconcelos, N.M.; Goncalves, A.; Vande Walle, L.; Demon, D.; Matusiak, M.; Van Hauwermeiren, F.; et al. Caspase-1 Engagement and TLR-Induced c-FLIP Expression Suppress ASC/Caspase-8-Dependent Apoptosis by Inflammasome Sensors NLRP1b and NLRC4. Cell Rep. 2017, 21, 3427–3444.
- Man, S.M.; Hopkins, L.J.; Nugent, E.; Cox, S.; Glück, I.M.; Tourlomousis, P.; Wright, J.A.; Cicuta, P.; Monie, T.P.; Bryant, C.E. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. USA 2014, 111, 7403–7408.
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515.
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Nunez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969.
- Gutierrez, K.D.; Davis, M.A.; Daniels, B.P.; Olsen, T.M.; Ralli-Jain, P.; Tait, S.W.; Gale, M., Jr.; Oberst, A. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1β Independently of Gasdermin-D. J. Immunol. 2017, 198, 2156–2164.
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897.
- Demarco, B.; Grayczyk, J.P.; Bjanes, E.; Le Roy, D.; Tonnus, W.; Assenmacher, C.A.; Radaelli, E.; Fettrelet, T.; Mack, V.; Linkermann, A.; et al. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci. Adv. 2020, 6, eabc3465.
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069.
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem. Biol. 2017, 24, 507–514.
- Platnich, J.M.; Chung, H.; Lau, A.; Sandall, C.F.; Bondzi-Simpson, A.; Chen, H.M.; Komada, T.; Trotman-Grant, A.C.; Brandelli, J.R.; Chun, J.; et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep. 2018, 25, 1525–1536.e1527.
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689.
- de Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161.
- Op de Beeck, K.; Van Camp, G.; Thys, S.; Cools, N.; Callebaut, I.; Vrijens, K.; Van Nassauw, L.; Van Tendeloo, V.F.; Timmermans, J.P.; Van Laer, L. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur. J. Hum. Genet. 2011, 19, 965–973.
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103.
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128.
- Zhou, B.; Abbott, D.W. Gasdermin E permits interleukin-1 beta release in distinct sublytic and pyroptotic phases. Cell Rep. 2021, 35, 108998.
- Wang, C.; Yang, T.; Xiao, J.; Xu, C.; Alippe, Y.; Sun, K.; Kanneganti, T.D.; Monahan, J.B.; Abu-Amer, Y.; Lieberman, J.; et al. NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci. Immunol. 2021, 6, eabj3859.
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Thi Nguyen, D.; Hattori, T.; Manh Le, T.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091.
- Heilig, R.; Dilucca, M.; Boucher, D.; Chen, K.W.; Hancz, D.; Demarco, B.; Shkarina, K.; Broz, P. Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci. Alliance 2020, 3, e202000735.
- Xu, W.; Che, Y.; Zhang, Q.; Huang, H.; Ding, C.; Wang, Y.; Wang, G.; Cao, L.; Hao, H. Apaf-1 Pyroptosome Senses Mitochondrial Permeability Transition. Cell Metab. 2021, 33, 424–436.e410.
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019, 38, e101638.
- Yan, W.T.; Yang, Y.D.; Hu, W.M.; Ning, W.Y.; Liao, L.S.; Lu, S.; Zhao, W.J.; Zhang, Q.; Xiong, K. Do pyroptosis, apoptosis, and necroptosis (PANoptosis) exist in cerebral ischemia? Evidence from cell and rodent studies. Neural Regen. Res. 2022, 17, 1761–1768.
- Jiang, W.; Deng, Z.; Dai, X.; Zhao, W. PANoptosis: A New Insight Into Oral Infectious Diseases. Front. Immunol. 2021, 12, 789610.
- Chi, D.; Lin, X.; Meng, Q.; Tan, J.; Gong, Q.; Tong, Z. Real-Time Induction of Macrophage Apoptosis, Pyroptosis, and Necroptosis by Enterococcus faecalis OG1RF and Two Root Canal Isolated Strains. Front. Cell. Infect. Microbiol. 2021, 11, 720147.
- Lin, J.F.; Hu, P.S.; Wang, Y.Y.; Tan, Y.T.; Yu, K.; Liao, K.; Wu, Q.N.; Li, T.; Meng, Q.; Lin, J.Z.; et al. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct. Target. Ther. 2022, 7, 54.
- Song, M.; Xia, W.; Tao, Z.; Zhu, B.; Zhang, W.; Liu, C.; Chen, S. Self-assembled polymeric nanocarrier-mediated co-delivery of metformin and doxorubicin for melanoma therapy. Drug Deliv. 2021, 28, 594–606.
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297.
- Kaiser, W.J.; Upton, J.W.; Mocarski, E.S. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 2008, 181, 6427–6434.
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 2016, 20, 674–681.
- Wang, Y.; Kanneganti, T.D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput. Struct. Biotechnol. J. 2021, 19, 4641–4657.
- Tamura, T.; Ishihara, M.; Lamphier, M.S.; Tanaka, N.; Oishi, I.; Aizawa, S.; Matsuyama, T.; Mak, T.W.; Taki, S.; Taniguchi, T. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature 1995, 376, 596–599.
- Tanaka, N.; Ishihara, M.; Kitagawa, M.; Harada, H.; Kimura, T.; Matsuyama, T.; Lamphier, M.S.; Aizawa, S.; Mak, T.W.; Taniguchi, T. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell 1994, 77, 829–839.
- Karki, R.; Kanneganti, T.D. The ‘cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705.
- Man, S.M.; Karki, R.; Malireddi, R.K.; Neale, G.; Vogel, P.; Yamamoto, M.; Lamkanfi, M.; Kanneganti, T.D. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol. 2015, 16, 467–475.


