Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Weronika Stróżewska | -- | 2710 | 2022-05-27 14:41:30 | | | |
| 2 | Nora Tang | -27 word(s) | 2683 | 2022-05-30 04:52:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Stróżewska, W.; Durda-Masny, M.; Szwed, A. Mutations in GHR and IGF1R Genes. Encyclopedia. Available online: https://encyclopedia.pub/entry/23497 (accessed on 25 July 2026).
Stróżewska W, Durda-Masny M, Szwed A. Mutations in GHR and IGF1R Genes. Encyclopedia. Available at: https://encyclopedia.pub/entry/23497. Accessed July 25, 2026.
Stróżewska, Weronika, Magdalena Durda-Masny, Anita Szwed. "Mutations in GHR and IGF1R Genes" Encyclopedia, https://encyclopedia.pub/entry/23497 (accessed July 25, 2026).
Stróżewska, W., Durda-Masny, M., & Szwed, A. (2022, May 27). Mutations in GHR and IGF1R Genes. In Encyclopedia. https://encyclopedia.pub/entry/23497
Stróżewska, Weronika, et al. "Mutations in GHR and IGF1R Genes." Encyclopedia. Web. 27 May, 2022.
Copy Citation
The birth size of a newborn child is influenced by a number of factors. The main ones are genetic factors of the fetus and the intrauterine environment. These factors interact with each other, and the effect of this interaction is seen as the birth weight, length, body composition, and organ size. From conception to delivery, the fetus is under the influence of the mother’s organism, which is the environment for the developing organism. The capacity of the uterus corresponds to the mother’s height and it is one of the main determinants of the fetus’s size. In addition, the birth size of the fetus can be influenced by the nutritional status of the mother, which provides nutrients to developing organisms.
growth hormone receptor
insulin-like growth factor 1 receptor
SGA
catch-up growth
1. Growth Hormone Receptor
The GHR gene is located on the 5p13.1-p12 chromosome and it is approximately 87 kbp. GHR is encoded by nine exons, the coding region, and a 3’ untranslated region. Eight of these exons are between 66 and 179 bp in size, with the last one, which encodes almost the entire cytoplasmic domain, being approximately 3400 bp [1].
Two isoforms of GHR have been distinguished in humans. Full-length isoform (fl-GHR) and an isoform that is lacking exon 3 (d3-GHR). By comparing the two isoforms in vitro, after the exposure to GH, it was found that the d3-GHR polymorphism has 1,7 to 2,0 times faster signal transduction than the full-length isoform. An association has also been noted between d3-GHR and an increased response to GH therapy in SGA children [2][3]. The largest number of GHRs is found in the liver; however, they are also found in other tissues and organs, such as the growth plate adipose tissue, muscle, and kidneys [4].
The growth process is initiated by binding the growth hormone to the GHR, which activates the intracellular signaling path through dimerization. GHR-related intracellular Janus tyrosine kinase 2 (JAK2) and, among others, the transcription activator 5 (STAT5), affect the expression of the IGF-1 gene, which results in the secretion of IGF-1. After the IGF-1 binds itself to IGF1R, this complex activates a mitogenic and anabolic response in target cells that lead to growth [5]. The extracellular domain of the GHR corresponds to GHBP, which is complexed with about half of the GH in human plasma [6].
Mutations in the GHR gene cause growth hormone insensitivity syndrome (Laron’s syndrome), as well as partial growth hormone sensitivity [7]. As of today, 26 mutations that are responsible for Laron’s syndrome have been discovered: one that is responsible for hypercholesterolemia, one for increased responsiveness to GH, and five that are responsible for partial sensitivity to growth hormone. The latter, which are the aim of this study, are presented in Figure 1.
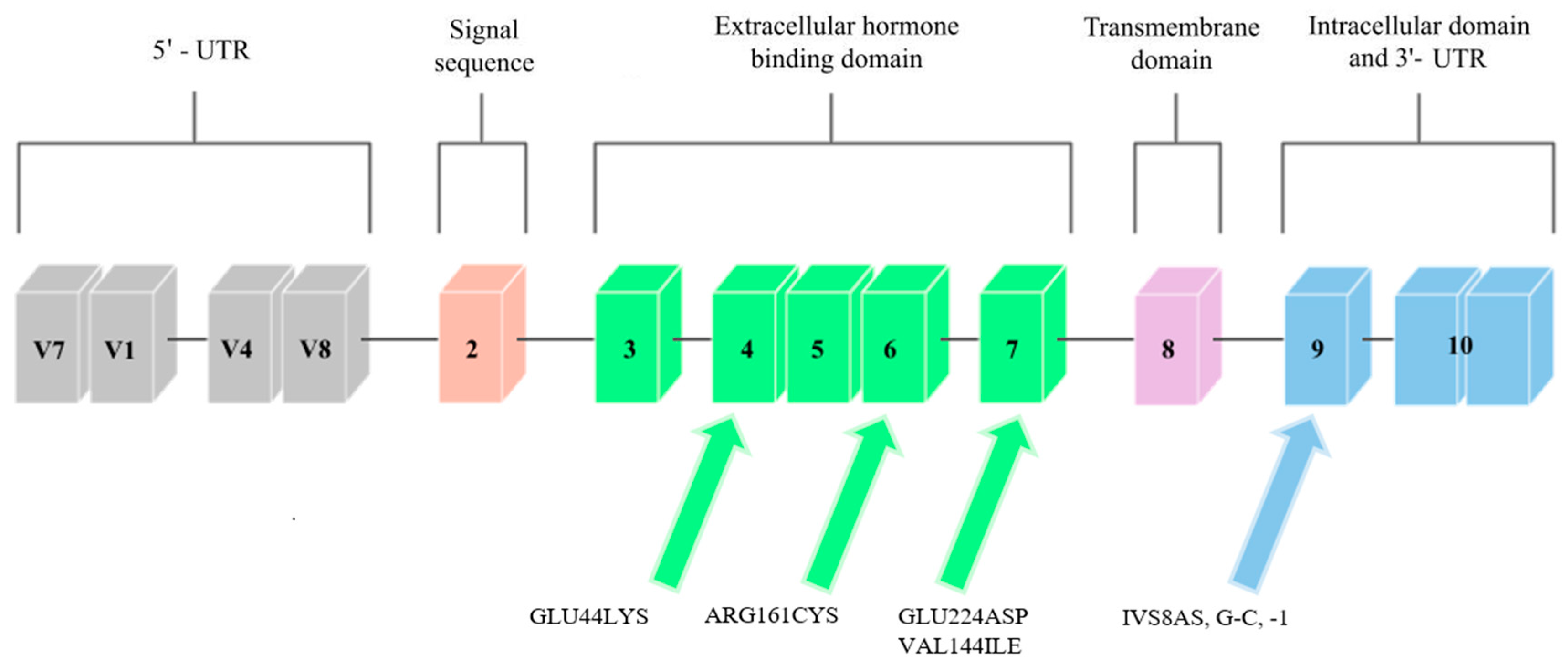
Figure 1. Scheme of the GHR gene. Arrows point to exons where mutations related to low body height or SGA/IUGR defined at birth have been found.
Three mutations that cause partial GH sensitivity in the GHR gene were discovered by Goddard et al. [8] in 4 out of 14 children with idiopathic short statures. The subjects were selected on the basis of the normal levels of growth-hormone secretion and the low serum levels of GH-binding protein.
Most of the previously identified mutations were located in the extracellular domain of the GHR, which, consequently, led to a decrease in the level of serum GHBP (which is derived from this receptor region). The results of the study by Goddard et al. [8] suggest that heterozygous mutations in the GHR gene may account for approximately 5% of idiopathic short-stature patients.
2. Insulin-like Growth Factor-1 Receptor
The IGF1R gene is located on chromosome 15q.25–q.26 [9]. It covers approximately 100 kb, and the coding sequence of this gene is contained in 21 exons. Cooke et al. [10] analyzed the promoter region and found that the flanking and untranslated region is rich in guanine–cytosine pairs. The diagram of this gene is shown in Figure 2.
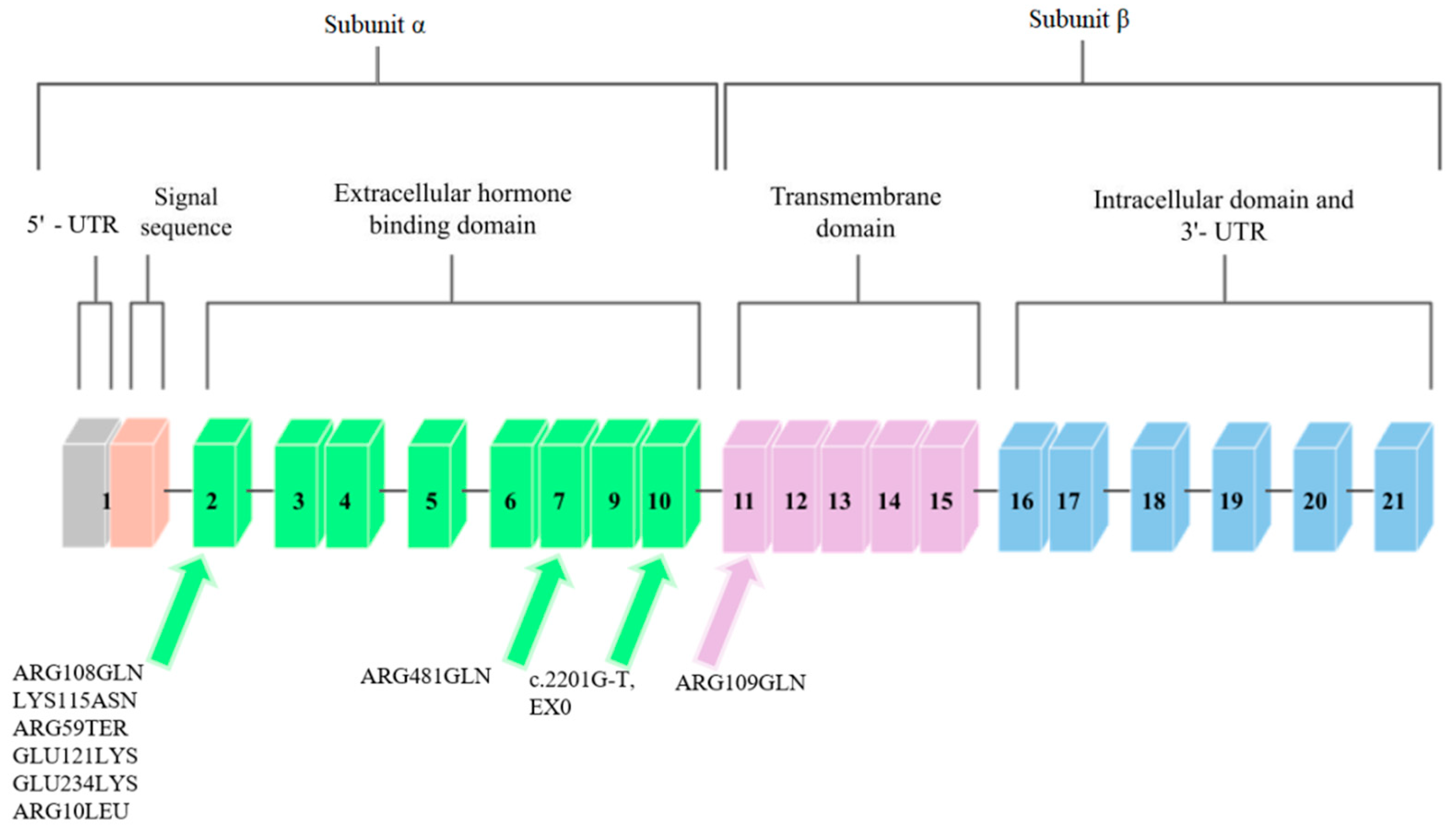
Figure 2. Schematic diagram of the IGF1R gene. Arrows point to exons where mutations related to low body height or SGA/IUGR defined at birth have been found.
IGF1R is homologous to the insulin receptor gene with regard to amino acid sequences, which are more than 50% identical, and with regard to the organization of introns and exons. These genes code the precursor proteins that are post-translationally modified to produce receptors that consist of two α subunits that are extracellular and that contain ligand-binding domains and two β that contain intracellular tyrosine kinase domains. The complete absence of IGF-1 receptors may cause severe disease or may be lethal in humans, and less severe disorders, such as naturally occurring nonsense mutations, result in insulin resistance and IGF-1 resistance [11].
IGF1R shows a high level of homology with the insulin receptor, and it activates comparable signal transduction pathways. IGF1R can also form hybrid receptors with the insulin receptor [12]. Ligand-IGF1R binding leads to the autophosphorylation of this receptor and to tyrosine phosphorylation. Subsequently, substrate phosphorylation activates two major signaling pathways, which are PI3K/AKT/mTOR and Ras-MAPK [13].
Mutations in the IGF1R gene can confer IGF-1 resistance and can, hence, cause some cases of prenatal and postnatal growth restriction [11]. Nine mutations in the IGF1R gene have been described so far. They are presented in Figure 2.
A case of a person with a chromosome 15q26.1-qter deletion and monozygotic IGF1R gene was described. The patient exhibited Intrauterine Growth Restriction (IUGR), delayed growth and development, and, inter alia, microcephaly, lung underdevelopment, and kidney abnormalities. On the basis of other patients with similar chromosome 15 deletions, the study authors suggest that the absence of the IGF1R allele may be associated with the development of IUGR and postnatal growth deficiency [14].
3. Mutations in the GHR Gene
3.1. GLU44LYS, rs121909361
One of the mutations that was diagnosed by Goddard et al. [8] was a mutation located in exon 4 that introduced lysine instead of glutamic acid at position 44 (Glu44Lys) in 184 base pairs. The mutation in this exon was inherited from the father. The authors of the study analyzed the effect of this mutation on the GH-binding ability of GHR. The Glu44 residue is in direct contact with the GH, and the introduction of lysine at position 44 reduced the binding by 330, compared to the wild-type receptor binding. It was therefore found that this mutation is responsible for a decreased affinity for GH.
3.2. ARG161CYS, rs121909362
Another mutation that was reported by Goddard et al. [8] was a mutation in exon 6, at 535 base pairs (Figure 1). It introduced a cysteine at position 161 (Arg161Cys) in place of arginine. The cysteine in this mutation was potentially misfolded. The mutation was inherited from the mother’s side. One of the patients studied by Goddard was heterozygously composed of the Glu44Lys and Arg161Cys mutations, and he was more affected by partial GH sensitivity than his homozygous parents (father had the Glu44Lys mutation, mother had Arg161Cys). Similar to the rest of the study participants, he was treated with GH, but the response to this treatment was poor, and so it was concluded that the reason for this may be mutations that cause partial sensitivity to GH due to dysfunctional GHRs.
3.3. GLU224ASP, rs45588036
The last mutation discovered by Goddard et al. [8] was found in exon 7 (Figure 1), and it was a mutation in the 726 base pair that introduced aspartic acid in place of glutamic acid at position 224 (Glu224Asp). However, the conservative substitution in this mutation was expressed at normal expression levels and resembled the mutant Arg211His protein, so that the GH affinity for the GHR gene that contains the Glu224Asp mutation was nearly normal. A possible effect of this mutation was incorrect subcellular localization. Partial GH insensitivity, which can be manifested by short stature, may occur in individuals who have heterozygous mutations in the GHR gene due to mutations in other genes, or when changes confer a dominant phenotype. Patients were selected, in part, on the basis of decreased serum GHBP levels, which suggest that the mutations discussed by Goddard may directly affect ligand binding. In the case of the Glu44Lys mutation, a 330-fold decrease in the affinity of the GH receptor to GHBP was observed, while the remaining mutations had smaller effects on the ligand binding.
3.4. IVS8AS, G-C, -1 rs730880308
Ayling et al. [15] report the case of a family in which a GHIS causal mutation, IVS8AS, was recognized in the mother and daughter, which was located in the cytoplasmic domain of the GH receptor, which is an inherited domain. It was a single heterozygous mutation that transverses G to C at position 1 of the 3′ splicing acceptor site that precedes exon 9 of the GHR (Figure 1). The fusion of JAK2 with the GHR requires a conserved exon 9-encoded sequence that is located in the cytoplasmic domain. This mutation had a dominant negative effect, which caused a shortening of the cytoplasmic part of the protein, and it was associated with GHBP. Most of the previously discovered GHR mutations were located in the extracellular binding domain of the GHR. However, in the study, Ayling indicates that dominant GHR mutations should be looked for in children with normal GHBP levels and low body heights; that is, in children with no previous suspected endocrinopathy.
3.5. VAL144ILE, rs6413484
A study by Sanchez et al. [16] discovered the last known mutation in the growth hormone receptor gene so far, which was the VAL144ILE mutation, which is located on exon 6. It was found in one subject with a short body height and in two members of her family, who also had shorter body heights (Figure 1). A study by Amslem et al. [7] describes the same amino acid that is changed in this family. The significance of this mutation has not been fully established; however, it seems likely that this mutation affects height. The potential significance of this mutation was established from the above-described work by Ayling et al. [15], in which the patient with GHIS had a mutated allele on the same amino acid (144) as the patient in the study by Sanchez et al. [16]. However, this mutation occurred in a different base pair. This change was related to GHIS, and so the amino acid V144 was found to be important for the GH receptor function.
4. Mutations in the IGF1R Gene
4.1 ARG108GLN, rs121912426
Abuzzahab et al. [11], after examining 51 people who showed short postnatal growth, found a mutation in the IGF1R gene in two of them. Both of these mutations were found in exon 2 (Figure 2). The first person described had two mutations. The first was a single base pair substitution at the codon of amino acid 108 that changed arginine into glutamine. The mutation at position 108, ARG108GLN, was inherited from the father. This mutation affected the α and β cleavage site, which resulted in the intracellular sequestration of the unprocessed proreceptor, and decreased IGF1R expression [17].
4.2 LYS115ASN, rs121912427
The second mutation described by Abuzzahab et al. [24] in the first of 51 patients was a mutation in IGF1R that consisted of replacing lysine with asparagine at amino acid codon 115 (Figure 2). This mutation, LYS115ASN, was inherited from the mother. In the person who had these mutations, each of the α subunits in the mature receptor contained one of these mutations in various combinations, which is due to the heterotetrameric structure of IGF1R. These mutations changed the conformation of the IGF1R-binding domain and, consequently, reduced ligand binding. The binding of IGF-1 to IGF1R on the fibroblasts in the patient was reduced by one-third compared to the control group. Receptor phosphory-lation in response to IGF-1 was also investigated to determine decreased hormone binding to receptor signaling. Phosphorylation was dose-dependent in fibroblasts; however, it showed a decrease in sensitivity compared to the control group. The subject was treated with growth hormone, during which the concentration of IGF-1 in the serum increased, which indicated abnormal GH secretion.
4.3 ARG59TER, rs121912428
Abuzzahab et al. [11] also found mutations in a patient who showed a low body height and a slow growth rate. This patient was heterozygous for the early transcriptional termination mutation at exon 2 at position 59, which should contain arginine (Figure 2). This mutation was inherited from the mother, who was also born SGA. This mutation inactivates one IGF1R allele, and the protein may not be expressed; however, if expressed, it would only correspond to the N-terminal end of the IGF1R α chain, and the receptor would not be able to bind IGF-1. It was suggested that individuals with the ARG59TER mutation may exhibit a haploinsufficiency phenotype, as the biallelic expression of wild-type and mutant IGF1R alleles was detected in the blood of the patient. After examining the phosphorylation of Akt, the IGF1R signaling protein, and following an analysis of the post-receptor signal transduction, it was proposed that the IGF-1 resistance in the subject was due to a reduced number of IGF1R [17].
4.4 ARG709GLN, rs121012429
The ARG709GLN mutation was discovered by Kawashima et al. [18]. They examined 24 people who were characterized by low body heights and IUGR. One person was identified with a mutation in the IGF1R gene that was heterozygous for arginine to glutamine conversion at position 709 (Figure 2). This mutation was inherited from the mother and it changed the IGF1R proreceptor cleavage site by losing the restriction site. ARG709GLN affected the cleavage site between the α and β subunits, which caused the intracellular sequestration of the unprocessed proreceptor, and the decreased expression of fully digested and processed IGF1R [17].
4.5 ARG481GLN, rs33958176
In another study, Inagaki et al. [19] describes a mutation that was found in a girl with a short body height. This mutation was based on the replacement of arginine with glutamine at position 481 of exon 7 IGF1R (Figure 2). This mutation was inherited from the maternal side. Researchers observed an increase in tyrosine phosphorylation after IGF-1 stimulation in the mutant IGF1R, compared to wild-type, and decreases in Akt phosphorylation and in proliferation in cells that overexpressed mutant IGF1R. Because of the mutation site being close to the first of the four disulfide bonds that connect the α and β subunits, it was suggested that this mutation disrupted this binding and resulted in incomplete dimerization and conformational alteration.
4.6. GLU121LYS, rs1555434208
The GLU121LYS and GLU234LYS mutations were discovered and described in the study by Fang et al. [20]. Two siblings were examined, one of whom had IUGR, and the other who had a short body height. Subjects had complex heterozygous missense mutations in the IGF1R gene. One was found in exon 2 and consisted of replacing glutamic acid with lysine at position 121 (Figure 2). This mutation was also found in the mother of the studied siblings.
4.7. GLU234LYS, rs1253103806
The second mutation reported by Fang et al. [20] is the mutation that is involved in replacing glutamic acid with lysine at position 234 of exon 2 (Figure 2). This mutation was inherited from the father of the subjects. These mutations resulted in decreased expression of mature and functional IGF1R peptides, with a consequently decreased activation of the IGF1R pathway in response to IGF-1. Analysis of sibling fibroblasts showed an 80% reduction in processed α and β subunits, compared to wild-type receptors.
4.8. ARG10LEU, rs1409058783
The penultimate mutation identified so far in the IGF1R gene was discovered and described by Gannage-Yared et al. [12]. They examined a patient born with IUGR who exhibited low body height later in life. A missense mutation in exon 2 was identified to convert arginine to leucine at position 10 (Figure 2). This mutation was inherited from both parents, who were its heterozygous carriers. This mutation resulted in the loss of hydrogen bonds between Leu10 and Asp8 in the IGF1R. The total expression of IGF1R containing this mutation was unchanged; however, impaired IGF receptor activation and decreased IGF1R autophosphorylation were observed in the subject’s fibroblasts, and greater amounts of IGF-1 were required to achieve a similar level of Akt phosphorylation as in the controls with receptors without this mutation.
4.9. EX10, rs1555460945, c.2201G-T
The most recent mutation in the IGF1R gene to date is reported by Prontera et al. [13]. The mutation carrier was a patient born with IUGR. The mutation in this person consisted of a c.2201G-T transversion that was identified on the last nucleotide in exon 10 (Figure 2). The examined person showed reduced autophosphorylation in fibroblasts and lower Akt activity, and an abnormal isoform that generates a mutant protein that contains 25 additional amino acids. The studies carried out by the authors of the study indicate the impaired ability of the IGF1R that contains this mutation to self-phosphorylate and to activate the downstream IGF-1 signaling pathway and the insulin–Akt complex.
References
- Godowski, P.; Leung, D.; Meacham, L.; Galgani, J.; Hellmiss, R.; Keret, R.; Rotwein, P.; Parks, J.; Laron, Z.; Wood, W. Characterization of the human growth hormone receptor gene and demonstration of a partial gene deletion in two patients with Laron-type dwarfism. Med. Sci. 1989, 86, 8083–8087.
- Dos Santos, C.; Essioux, L.; Teinturier, C.; Tauber, M.; Goffin, V.; Bougnères, P. A common polymorphism of the growth hormone receptor is associated with increased responsiveness to growth hormone. Nat. Genet. 2004, 36, 720–724.
- Wegmann, M.; Thankamony, A.; Roche, E.; Hoey, H.; Kirk, J.; Shaikh, G.; Ivarsson, S.; Söder, O.; Dunger, D.; Juul, A.; et al. The exon3-deleted growth hormone receptor gene polymorphism (d3-GHR) is associated with insulin and spontaneous growth in short SGA children (NESGAS). Growth Horm. IGF Res. 2017, 35, 45–51.
- Dehkhoda, F.; Lee, C.M.M.; Medina, J.; Brooks, A.J. The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects. Front. Endocrinol. 2018, 9, 35.
- Gan, Y.; Buckels, A.; Liu, Y.; Zhang, Y.; Paterson, A.; Jiang, J.; Zinn, K.; Frank, S. Human GH receptor-IGF-1 receptor interaction: Implications for GH signaling. Mol. Endocrinol. 2014, 28, 1841–1854.
- Amit, T.; Moussa, B.; Youdim, H.; Hochberg, Z. Does Serum Growth Hormone (GH) Binding Protein Reflect Human GH Receptor Function? J. Clin. Endocrinol. Metab. 2000, 85, 927–932.
- Amselem, S.; Sobrier, M.; Duquesnoy, P.; Rappaport, R.; Postel-Vinay, M.; Gourmelen, M.; Dallapiccola, B.; Goossens, M. Recurrent nonsense mutations in the growth hormone receptor from patients with Laron dwarfism. J. Clin. Investig. 1991, 87, 1098–1102.
- Goddard, D.; Covello, R.; Luoh, S.; Clackson, T.; Attie, K.; Gesundheit, N.; Rundle, A.; Wells, J.; Carlsson, L. Mutations of the Growth Hormone Receptor in Children with Idiopathic Short Stature. The Growth Hormone Insensitivity Study Group. N. Engl. J. Med. 1995, 333, 1093–1098.
- Francke, U.; Yang-Feng, T.; Brissenden, J.; Ullrich, A. Chromosomal mapping of genes involved in growth control. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 855–866.
- Cooke, D.; Bankert, L.; Roberts, C., Jr.; LeRoith, D.; Casella, S. Analysis of the human type I insulin-like growth factor receptor promoter region. Biochem. Biophys. Res. Commun. 1991, 177, 1113–1120.
- Abuzzahab, M.; Schneider, A.; Goddard, A.; Grigorescu, F.; Lautier, C.; Keller, E.; Kiess Wi Klammt, J.; Kratzsch, J.; Osgood, D.; Pfäffle, R.; et al. IGF-I Receptor Mutations Resulting in Intrauterine and Postnatal Growth Retardation. N. Engl. J. Med. 2003, 349, 2211–2222.
- Gannagé-Yared, M.; Klammt, J.; Chouery, E.; Corbani, S.; Mégarbané, H.; Abou, G.; Choucair, N.; Pfäffle, R.; Mégarbané, A. Homozygous mutation of the IGF1 receptor gene in a patient with severe pre- and postnatal growth failure and congenital malformations. Eur. J. Endocrinol. 2013, 168, K1–K7.
- Prontera, P.; Micale, L.; Verrotti, A.; Napolioni, V.; Stangoni, G.; Merla, G. A New Homozygous IGF1R Variant Defines a Clinically Recognizable Incomplete Dominant form of SHORT Syndrome. Hum. Mutat. 2015, 36, 1043–1047.
- Roback, E.; Barakat, A.; Dev, V.; Mbikay, M.; Chretien, M.; Butler, M. An infant with deletion of the distal long arm of chromosome 15 (q26.1----qter) and loss of insulin-like growth factor 1 receptor gene. Am. J. Med. Genet. 1991, 38, 74–79.
- Ayling, R.; Ross, R.; Towner, P.; Von Laue, S.; Finidori, J.; Moutoussamy, S.; Buchanan, C.; Clayton, P.; Norman, M. A dominant-negative mutation of the growth hormone receptor causes familial short stature. Nat. Genet. 1997, 16, 13–14.
- Sanchez, J.; Perera, E.; Baumbach, L.; Cleveland, W. Growth Hormone Receptor Mutations in Children with Idiopathic Short Stature. J. Clin. Endocrinol. Metab. 1998, 83, 4079–4083.
- Raile, K.; Klammt, J.; Schneider, A.; Keller, A.; Laue, S.; Smith, R.; Pfäffle, R.; Kratzsch, J.; Keller, E.; Kiess, W. Clinical and functional characteristics of the human Arg59Ter insulin-like growth factor i receptor (IGF1R) mutation: Implications for a gene dosage effect of the human IGF1R. J. Clin. Endocrinol. Metab. 2006, 91, 2264–2271.
- Kawashima Yuki Kanzaki, S.; Yang, F.; Kinoshita, T.; Hanaki, K.; Nagaishi, J.; Ohtsuka, Y.; Hisatome, I.; Ninomoya, H.; Nanba, E.; Fukushima, T.; et al. Mutation at cleavage site of insulin-like growth factor receptor in a short-stature child born with intrauterine growth retardation. J. Clin. Endocrinol. Metab. 2005, 90, 4679–4687.
- Inagaki, K.; Tiulpakov, A.; Rubtsov Petr Sverdlova, P.; Peterkova, V.; Yakar, S.; Terekhov, S.; LeRoith, D. A familial insulin-like growth factor-I receptor mutant leads to short stature: Clinical and biochemical characterization. J. Clin. Endocrinol. Metab. 2007, 92, 1542–1548.
- Fang, P.; Cho, Y.; Derr, M.; Rosenfeld, R.; Hwa, V.; Cowell, C. Severe short stature caused by novel compound heterozygous mutations of the insulin-like growth factor 1 receptor (IGF1R). J. Clin. Endocrinol. Metab. 2012, 97, 224–243.
More
Information
Subjects:
Developmental Biology; Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
30 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No