Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Weronika Stróżewska and Version 2 by Nora Tang.
The birth size of a newborn child is influenced by a number of factors. The main ones are genetic factors of the fetus and the intrauterine environment. These factors interact with each other, and the effect of this interaction is seen as the birth weight, length, body composition, and organ size. From conception to delivery, the fetus is under the influence of the mother’s organism, which is the environment for the developing organism. The capacity of the uterus corresponds to the mother’s height and it is one of the main determinants of the fetus’s size. In addition, the birth size of the fetus can be influenced by the nutritional status of the mother, which provides nutrients to developing organisms.
- growth hormone receptor
- insulin-like growth factor 1 receptor
- SGA
- catch-up growth
1. Growth Hormone Receptor
The GHR gene is located on the 5p13.1-p12 chromosome and it is approximately 87 kbp. GHR is encoded by nine exons, the coding region, and a 3’ untranslated region. Eight of these exons are between 66 and 179 bp in size, with the last one, which encodes almost the entire cytoplasmic domain, being approximately 3400 bp [1][16].
Two isoforms of GHR have been distinguished in humans. Full-length isoform (fl-GHR) and an isoform that is lacking exon 3 (d3-GHR). By comparing the two isoforms in vitro, after the exposure to GH, it was found that the d3-GHR polymorphism has 1,7 to 2,0 times faster signal transduction than the full-length isoform. An association has also been noted between d3-GHR and an increased response to GH therapy in SGA children [2][3][17,18]. The largest number of GHRs is found in the liver; however, they are also found in other tissues and organs, such as the growth plate adipose tissue, muscle, and kidneys [4][9].
The growth process is initiated by binding the growth hormone to the GHR, which activates the intracellular signaling path through dimerization. GHR-related intracellular Janus tyrosine kinase 2 (JAK2) and, among others, the transcription activator 5 (STAT5), affect the expression of the IGF-1 gene, which results in the secretion of IGF-1. After the IGF-1 binds itself to IGF1R, this complex activates a mitogenic and anabolic response in target cells that lead to growth [5][19]. The extracellular domain of the GHR corresponds to GHBP, which is complexed with about half of the GH in human plasma [6][7].
Mutations in the GHR gene cause growth hormone insensitivity syndrome (Laron’s syndrome), as well as partial growth hormone sensitivity [7][20]. As of today, 26 mutations that are responsible for Laron’s syndrome have been discovered: one that is responsible for hypercholesterolemia, one for increased responsiveness to GH, and five that are responsible for partial sensitivity to growth hormone. The latter, which are the aim of this study, are presented in Figure 12.
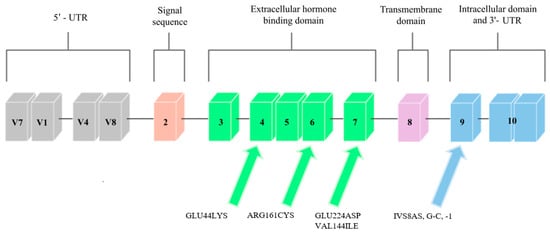
Figure 12. Scheme of the GHR gene. Arrows point to exons where mutations related to low body height or SGA/IUGR defined at birth have been found.
Three mutations that cause partial GH sensitivity in the GHR gene were discovered by Goddard et al. [8][21] in 4 out of 14 children with idiopathic short statures. The subjects were selected on the basis of the normal levels of growth-hormone secretion and the low serum levels of GH-binding protein.
Most of the previously identified mutations were located in the extracellular domain of the GHR, which, consequently, led to a decrease in the level of serum GHBP (which is derived from this receptor region). The results of the study by Goddard et al. [8][21] suggest that heterozygous mutations in the GHR gene may account for approximately 5% of idiopathic short-stature patients.
2. Insulin-like Growth Factor-1 Receptor
The IGF1R gene is located on chromosome 15q.25–q.26 [9][22]. It covers approximately 100 kb, and the coding sequence of this gene is contained in 21 exons. Cooke et al. [10][23] analyzed the promoter region and found that the flanking and untranslated region is rich in guanine–cytosine pairs. The diagram of this gene is shown in Figure 23.
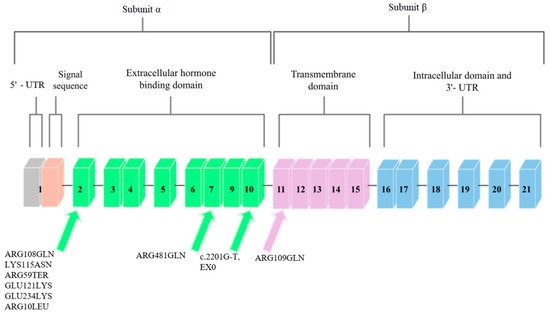
Figure 23. Schematic diagram of the IGF1R gene. Arrows point to exons where mutations related to low body height or SGA/IUGR defined at birth have been found.
IGF1R is homologous to the insulin receptor gene with regard to amino acid sequences, which are more than 50% identical, and with regard to the organization of introns and exons. These genes code the precursor proteins that are post-translationally modified to produce receptors that consist of two α subunits that are extracellular and that contain ligand-binding domains and two β that contain intracellular tyrosine kinase domains. The complete absence of IGF-1 receptors may cause severe disease or may be lethal in humans, and less severe disorders, such as naturally occurring nonsense mutations, result in insulin resistance and IGF-1 resistance [11][24].
IGF1R shows a high level of homology with the insulin receptor, and it activates comparable signal transduction pathways. IGF1R can also form hybrid receptors with the insulin receptor [12][25]. Ligand-IGF1R binding leads to the autophosphorylation of this receptor and to tyrosine phosphorylation. Subsequently, substrate phosphorylation activates two major signaling pathways, which are PI3K/AKT/mTOR and Ras-MAPK [13][26].
Mutations in the IGF1R gene can confer IGF-1 resistance and can, hence, cause some cases of prenatal and postnatal growth restriction [11][24]. Nine mutations in the IGF1R gene have been described so far. They are presented in Figure 23.
A case of a person with a chromosome 15q26.1-qter deletion and monozygotic IGF1R gene was described. The patient exhibited Intrauterine Growth Restriction (IUGR), delayed growth and development, and, inter alia, microcephaly, lung underdevelopment, and kidney abnormalities. On the basis of other patients with similar chromosome 15 deletions, the study authors suggest that the absence of the IGF1R allele may be associated with the development of IUGR and postnatal growth deficiency [14][27].