 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Roddy Hiram | -- | 2668 | 2022-05-26 17:41:39 | | | |
| 2 | Roddy Hiram | Meta information modification | 2668 | 2022-05-26 17:52:00 | | | | |
| 3 | Conner Chen | -20 word(s) | 2648 | 2022-05-27 04:31:58 | | |
Video Upload Options
Inflammation is a complex program of active processes characterized by the well-orchestrated succession of an initiation and a resolution phase aiming to promote homeostasis. When the resolution of inflammation fails, the tissue undergoes an unresolved inflammatory status which, if it remains uncontrolled, can lead to chronic inflammatory disorders due to aggravation of structural damages, development of a fibrous area, and loss of function. Various human conditions show a typical unresolved inflammatory profile. Inflammatory diseases include cancer, neurodegenerative disease, asthma, right heart disease, atherosclerosis, myocardial infarction, or atrial fibrillation. New evidence has started to emerge on the role, including pro-resolution involvement of chemical mediators in the acute phase of inflammation. Although flourishing knowledge is available about the role of specialized pro-resolving mediators in neurodegenerative diseases, atherosclerosis, obesity, or hepatic fibrosis, little is known about their efficacy to combat inflammation-associated arrhythmogenic cardiac disorders. It has been shown that resolvins, including RvD1, RvE1, or Mar1, are bioactive mediators of resolution. Resolvins can stop neutrophil activation and infiltration, stimulate monocytes polarization into anti-inflammatory-M2-macrophages, and activate macrophage phagocytosis of inflammation-debris and neutrophils to promote efferocytosis and clearance.
1. Introduction
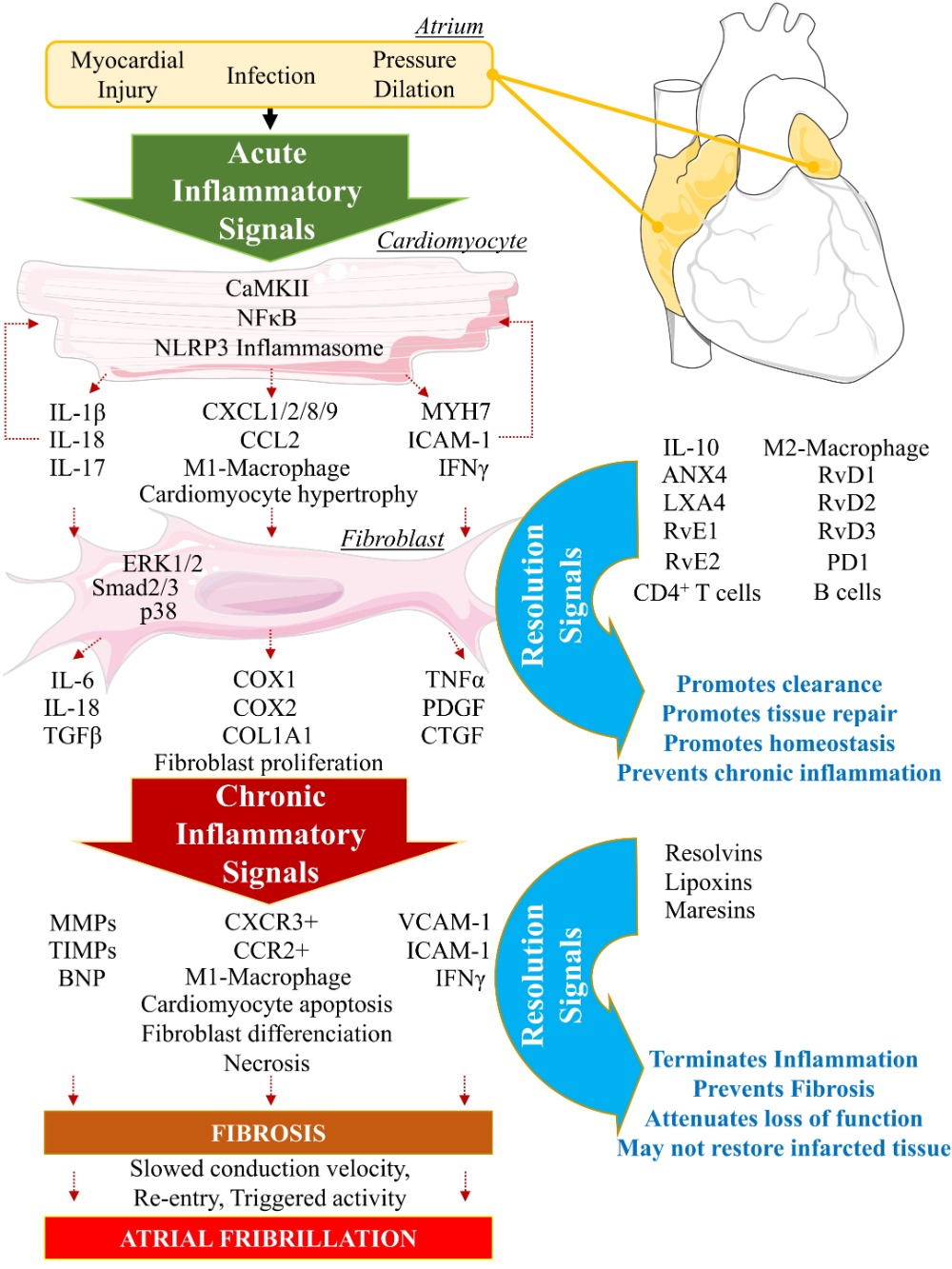
2. Biomolecular Paradigm of Active Resolution Mechanisms in the Heart
2.1. Initiation Phase of Inflammation: Central Regulatory Role of Arachidonic Acid
2.1.1. Arachidonic Acid Metabolism by Cytochrome P450
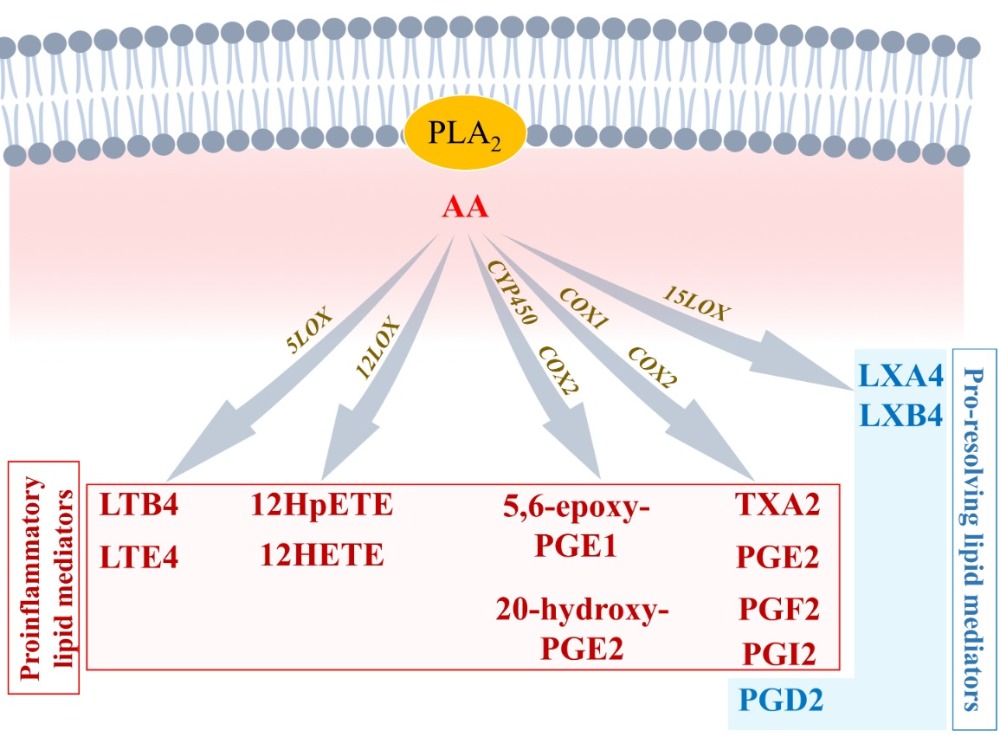
2.1.2. Arachidonic Acid Metabolism by COX1 and COX2
2.1.3. Arachidonic Acid Metabolism by 5-LOX
2.2. Lipid-Mediator Class Switching: Transition from Pro-Inflammatory to Pro-Resolution Signals
2.3. Resolution of Inflammation: SPMs-Mediated Efferocytosis and Homeostasis
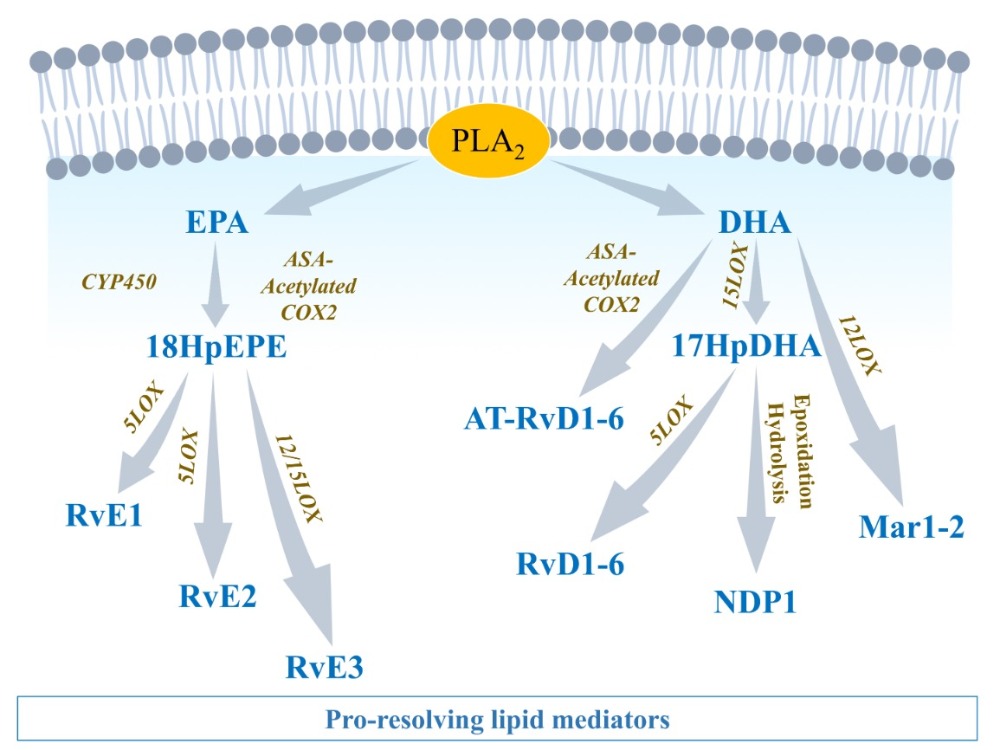
2.3.1. EPA-Derived Specialized Pro-Resolving Mediators
2.3.2. DHA-Derived Specialized Pro-Resolving Mediators
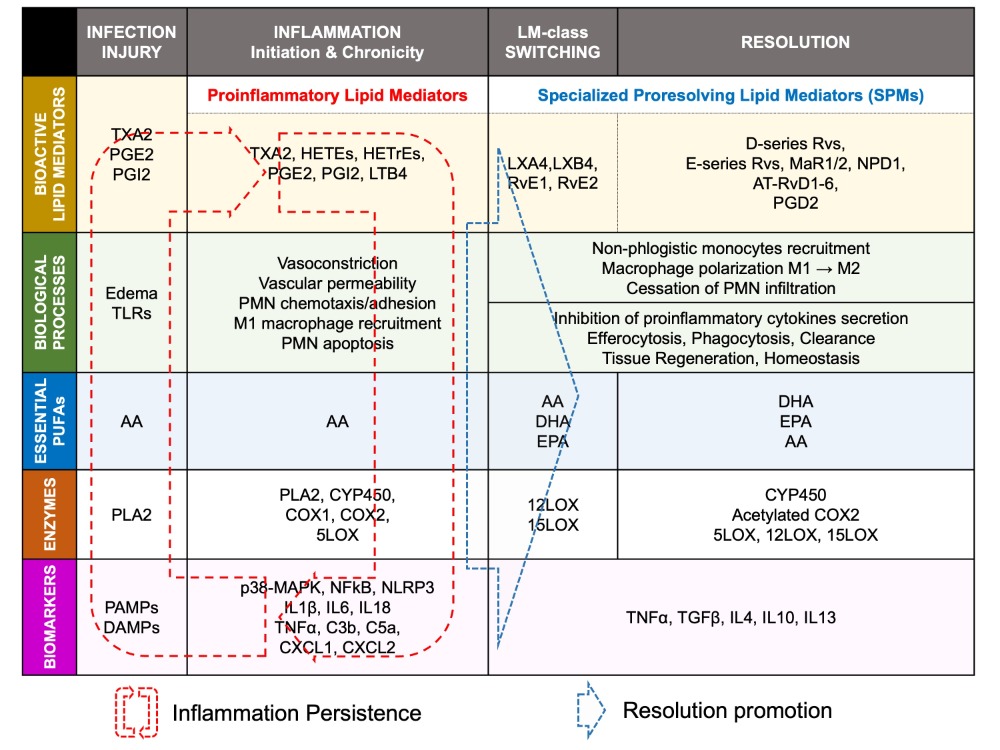
2.3.3. Arachidonic Acid-Derived Specialized Pro-Resolving Mediators
2.4. ‘Failed Resolution Mechanisms’ in the Development of Chronic Inflammation and Heart Diseases
References
- Andrade, J.; Khairy, P.; Dobrev, D.; Nattel, S. The clinical profile and pathophysiology of atrial fibrillation: Relationships among clinical features, epidemiology, and mechanisms. Circ. Res. 2014, 114, 1453–1468.
- Hu, Y.-F.; Chen, Y.-J.; Lin, Y.-J.; Chen, S.-A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243.
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of Inflammation in Atrial Fibrillation Pathophysiology and Management. Circ. J. 2015, 79, 495–502.
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship Between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338.
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112.
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 160.
- Sun, Z.; Zhou, D.; Xie, X.; Wang, S.; Wang, Z.; Zhao, W.; Xu, H.; Zheng, L. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res. Cardiol. 2016, 111, 63.
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2013, 70, 74–82.
- Frangogiannis, N.G. The immune system and cardiac repair. Pharmacol. Res. 2008, 58, 88–111.
- Whitehead, A.J.; Engler, A.J. Regenerative cross talk between cardiac cells and macrophages. Am. J. Physiol. Circ. Physiol. 2021, 320, H2211–H2221.
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis—Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250.
- Scott, L., Jr.; Li, N.; Dobrev, D. Role of inflammatory signaling in atrial fibrillation. Int. J. Cardiol. 2019, 287, 195–200.
- Hiram, R. Cardiac cytokine therapy? Relevance of targeting inflammatory mediators to combat cardiac arrhythmogenic remodeling. IJC Heart Vasc. 2021, 37, 100918.
- Akdis, D.; Chen, K.; Saguner, A.M.; Stämpfli, S.F.; Chen, X.; Chen, L.; Rao, M.; Haegeli, L.M.; Tanner, F.C.; Brunckhorst, C.; et al. Clinical Characteristics of Patients with a Right Ventricular Thrombus in Arrhythmogenic Right Ventricular Cardiomyopathy. Thromb. Haemost. 2019, 119, 1373–1378.
- Keramida, K.; Lazaros, G.; Nihoyannopoulos, P. Right ventricular involvement in hypertrophic cardiomyopathy: Patterns and implications. Hell. J. Cardiol. 2018, 61, 3–8.
- Tadic, M.; Cuspidi, C.; Pencic, B.; Sljivic, A.; Ivanovic, B.; Neskovic, A.; Scepanovic, R.; Celic, V. High-normal blood pressure impacts the right heart mechanics: A three-dimensional echocardiography and two-dimensional speckle tracking imaging study. Blood Press. Monit. 2014, 19, 145–152.
- Waligóra, M.; Tyrka, A.; Miszalski-Jamka, T.; Urbańczyk-Zawadzka, M.; Podolec, P.; Kopeć, G. Right atrium enlargement predicts clinically significant supraventricular arrhythmia in patients with pulmonary arterial hypertension. Heart Lung 2018, 47, 237–242.
- Scheel, P.J., III; Murray, B.; Tichnell, C.; James, C.A.; Tandri, H.; Calkins, H.; Chelko, S.P.; Gilotra, N.A. Arrhythmogenic Right Ventricular Cardiomyopathy Presenting as Clinical Myocarditis in Women. Am. J. Cardiol. 2021, 145, 128–134.
- Sun, X.-Q.; Abbate, A.; Bogaard, H.-J. Role of cardiac inflammation in right ventricular failure. Cardiovasc. Res. 2017, 113, 1441–1452.
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285, Erratum in Nat. Rev. Cardiol. 2021, 18, 735.
- Everett, T.H., IV; Olgin, J.E. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 2007, 4 (Suppl. 3), S24–S27.
- Harada, M.; Nattel, S. Implications of Inflammation and Fibrosis in Atrial Fibrillation Pathophysiology. Card. Electrophysiol. Clin. 2021, 13, 25–35.
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A (2) regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6.
- Sofogianni, A.; Alkagiet, S.; Tziomalos, K. Lipoprotein-associated Phospholipase A2 and Coronary Heart Disease. Curr. Pharm. Des. 2018, 24, 291–296.
- Madjid, M.; Ali, M.; Willerson, J.T. Lipoprotein-associated phospholipase A2 as a novel risk marker for cardiovascular disease: A systematic review of the literature. Tex. Heart Inst. J. 2010, 37, 25–39.
- Tjoelker, L.W.; Wilder, C.; Eberhardt, C.; Stafforinit, D.M.; Dietsch, G.; Schimpf, B.; Hooper, S.; Le Trong, H.; Cousens, L.S.; Zimmerman, G.A.; et al. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature 1995, 374, 549–553.
- Kroetz, D.L.; Zeldin, D. Cytochrome P450 pathways of arachidonic acid metabolism. Curr. Opin. Lipidol. 2002, 13, 273–283.
- Rossi, A.G.; O’Flaherty, J.T. Bioactions of 5-hydroxyicosatetraenoate and its interaction with platelet-activating factor. Lipids 1991, 26, 1184–1188.
- Marnett, L.J.; Rowlinson, S.W.; Goodwin, D.; Kalgutkar, A.S.; Lanzo, C.A. Arachidonic Acid Oxygenation by COX-1 and COX-2. J. Biol. Chem. 1999, 274, 22903–22906.
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704.
- Rådmark, O.; Samuelsson, B. 5-Lipoxygenase: Mechanisms of regulation. J. Lipid Res. 2009, 50, S40–S45.
- Maayah, Z.H.; El-Kadi, A.O.S. 5-, 12- and 15-Hydroxyeicosatetraenoic acids induce cellular hypertrophy in the human ventricular cardiomyocyte, RL-14 cell line, through MAPK- and NF-κB-dependent mechanism. Arch. Toxicol. 2015, 90, 359–373.
- Zhou, Y.; Khan, H.; Xiao, J.; Cheang, W.S. Effects of Arachidonic Acid Metabolites on Cardiovascular Health and Disease. Int. J. Mol. Sci. 2021, 22, 12029.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242.
- Pirault, J.; Bäck, M. Lipoxin and Resolvin Receptors Transducing the Resolution of Inflammation in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 1273.
- Recchiuti, A.; Mattoscio, D.; Isopi, E. Roles, Actions, and Therapeutic Potential of Specialized Pro-resolving Lipid Mediators for the Treatment of Inflammation in Cystic Fibrosis. Front. Pharmacol. 2019, 10, 252.
- Fiore, S.; Brezinski, M.E.; Sheppard, K.-A.; Serhan, C.N. The Lipoxin Biosynthetic Circuit and their Actions with Human Neutrophils. Adv. Exp. Med. Biol. 1991, 314, 109–132.
- Levy, B.D.; Clish, C.; ASchmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619.
- Reina-Couto, M.; Carvalho, J.; Valente, M.J.; Vale, L.; Afonso, J.; Carvalho, F.; Bettencourt, P.; Sousa, T.; Albino-Teixeira, A. Impaired resolution of inflammation in human chronic heart failure. Eur. J. Clin. Investig. 2014, 44, 527–538.
- Shi, Y.; Pan, H.; Zhang, H.-Z.; Zhao, X.-Y.; Jin, J.; Wang, H.-Y. Lipoxin A4 mitigates experimental autoimmune myocarditis by regulating inflammatory response, NF-κB and PI3K/Akt signaling pathway in mice. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1850–1859.
- Kain, V.; Liu, F.; Kozlovskaya, V.; Ingle, K.A.; Bolisetty, S.; Agarwal, A.; Khedkar, S.; Prabhu, S.D.; Kharlampieva, E.; Halade, G.V. Resolution Agonist 15-epi-Lipoxin A4 Programs Early Activation of Resolving Phase in Post-Myocardial Infarction Healing. Sci. Rep. 2017, 7, 9999.
- Ishihara, T.; Yoshida, M.; Arita, M. Omega-3 fatty acid-derived mediators that control inflammation and tissue homeostasis. Int. Immunol. 2019, 31, 559–567.
- Cairns, J.A. The coxibs and traditional nonsteroidal anti-inflammatory drugs: A current perspective on cardiovascular risks. Can. J. Cardiol. 2007, 23, 125–131.
- El Kebir, D.; Gjorstrup, P.; Filep, J.G. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 14983–14988.
- Serhan, C.N.; Arita, M.; Hong, S.; Gotlinger, K. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: An overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat. 2004, 73, 155–172.
- Duvall, M.G.; Levy, B.D. DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur. J. Pharmacol. 2015, 785, 144–155.
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arter. Thromb. Vasc. Biol. 2011, 31, 986–1000.
- Oni-Orisan, A.; Alsaleh, N.; Lee, C.R.; Seubert, J.M. Epoxyeicosatrienoic acids and cardioprotection: The road to translation. J. Mol. Cell. Cardiol. 2014, 74, 199–208.
- Serhan, C.N.; Gupta, S.K.; Perretti, M.; Godson, C.; Brennan, E.; Li, Y.; Soehnlein, O.; Shimizu, T.; Werz, O.; Chiurchiù, V.; et al. The Atlas of Inflammation Resolution (AIR). Mol. Asp. Med. 2020, 74, 100894.
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832.
- Qu, X.; Zhang, X.; Yao, J.; Song, J.; Nikolic-Paterson, D.J.; Li, J. Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. J. Pathol. 2012, 228, 506–519.
- Kain, V.; Ingle, K.A.; Colas, R.A.; Dalli, J.; Prabhu, S.D.; Serhan, C.N.; Joshi, M.D.; Halade, G.V. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J. Mol. Cell. Cardiol. 2015, 84, 24–35.
- Hiram, R.; Xiong, F.; Naud, P.; Xiao, J.; Sirois, M.; Tanguay, J.-F.; Tardif, J.-C.; Nattel, S. The inflammation-resolution promoting molecule resolvin-D1 prevents atrial proarrhythmic remodelling in experimental right heart disease. Cardiovasc. Res. 2020, 117, 1776–1789.

