Inflammation is a complex program of active processes characterized by the well-orchestrated succession of an initiation and a resolution phase aiming to promote homeostasis. When the resolution of inflammation fails, the tissue undergoes an unresolved inflammatory status which, if it remains uncontrolled, can lead to chronic inflammatory disorders due to aggravation of structural damages, development of a fibrous area, and loss of function. Various human conditions show a typical unresolved inflammatory profile. Inflammatory diseases include cancer, neurodegenerative disease, asthma, right heart disease, atherosclerosis, myocardial infarction, or atrial fibrillation.
New evidence has started to emerge on the role, including pro-resolution involvement of chemical mediators in the acute phase of inflammation. Although flourishing knowledge is available about the role of specialized pro-resolving mediators in neurodegenerative diseases, atherosclerosis, obesity, or hepatic fibrosis, little is known about their efficacy to combat inflammation-associated arrhythmogenic cardiac disorders. It has been shown that resolvins, including RvD1, RvE1, or Mar1, are bioactive mediators of resolution. Resolvins can stop neutrophil activation and infiltration, stimulate monocytes polarization into anti-inflammatory-M2-macrophages, and activate macrophage phagocytosis of inflammation-debris and neutrophils to promote efferocytosis and clearance.
This review aims to discuss the paradigm of failed-resolution mechanisms (FRM) potentially promoting arrhythmogenicity in right heart disease-induced inflammatory status.
- inflammation
- right heart disease
- atrial fibrillation
- resolution
- arrhythmias
- specialized pro-resolving mediators
- resolvins
1. Introduction
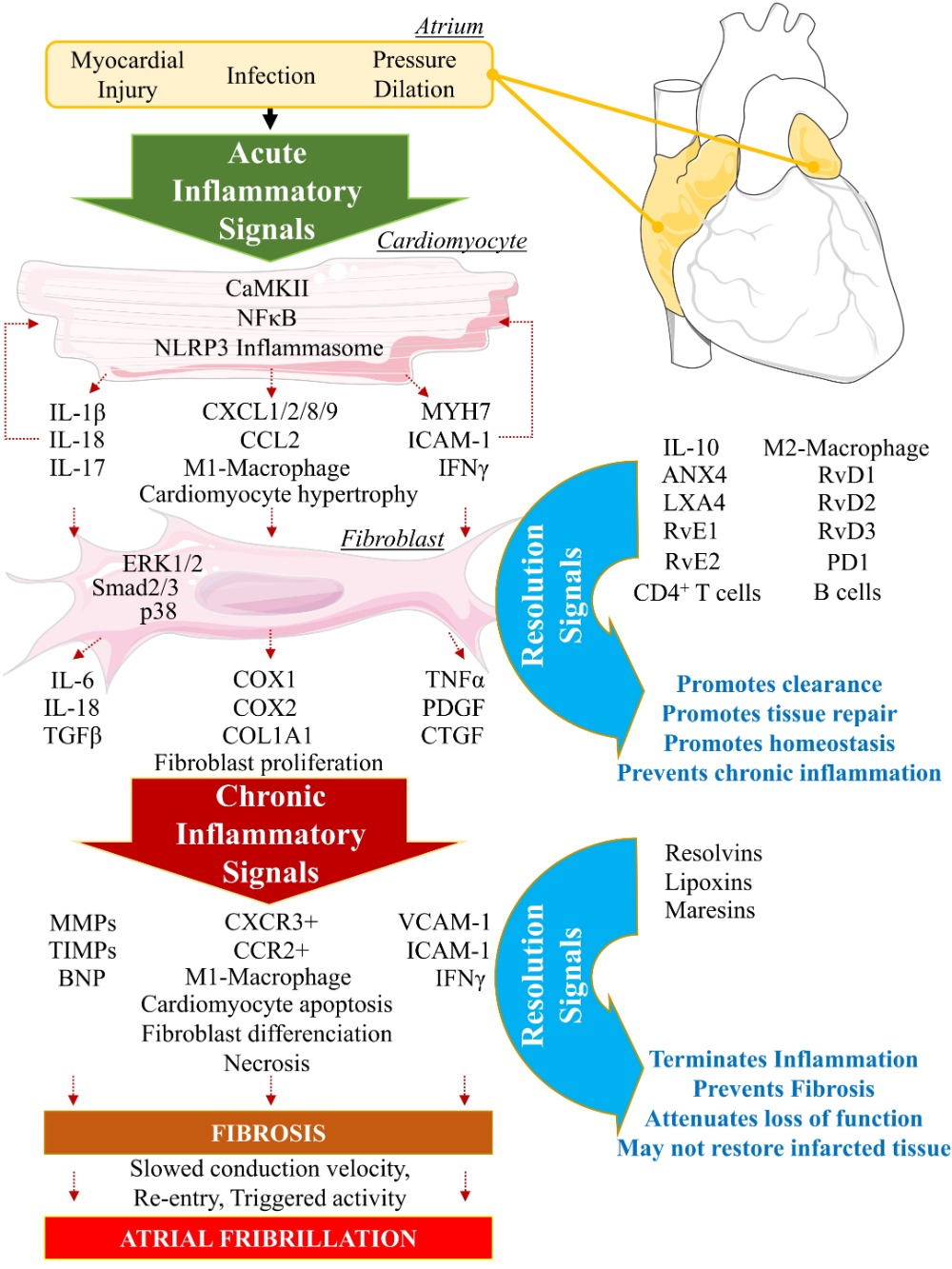
2. Biomolecular Paradigm of Active Resolution Mechanisms in the Heart
2.1. Initiation Phase of Inflammation: Central Regulatory Role of Arachidonic Acid
During the initiation of acute inflammation, phospholipase A2 (PLA2) levels are increased at the site of injury [23]. PLA2 produces arachidonic acid (AA: 5, 8, 11, 14-eicosatetraenoic acid) by hydrolyzation of the sn-2 ester bond of cellular phospholipids [23]. Patients with coronary artery disease show increased levels of lipoprotein-associated PLA2 (Lp-PLA2) [24]. Elevated levels of Lp-PLA2 have been suggested as an important risk factor of cardiovascular diseases [25]. Paradoxically, when Lp-PLA2 hydrolyzes the platelet-activating factor (PAF), its enzymatic activity is associated with anti-inflammatory properties [26]. The underlying mechanisms governing this paradox will be discussed below.2.1.1. Arachidonic Acid Metabolism by Cytochrome P450
AA is an essential polyunsaturated fatty acid (omega-6 PUFA) that can interact with cytochrome P450 (CYP450) enzymes to undergo monooxygenation or epoxidation and produce hydroxyeicosatetraenoic acids (19- and 20-HETEs) and dihydroxyeicosatrienoic acid (diHETrEs) [27] (Figure 2). These molecules act as hormone-like autocrine and paracrine agents to promote vasoconstriction, vascular permeability, polymorphonuclear leukocytes (PMN), and proinflammatory (M1)-macrophages chemotaxis, and proinflammatory signaling [28].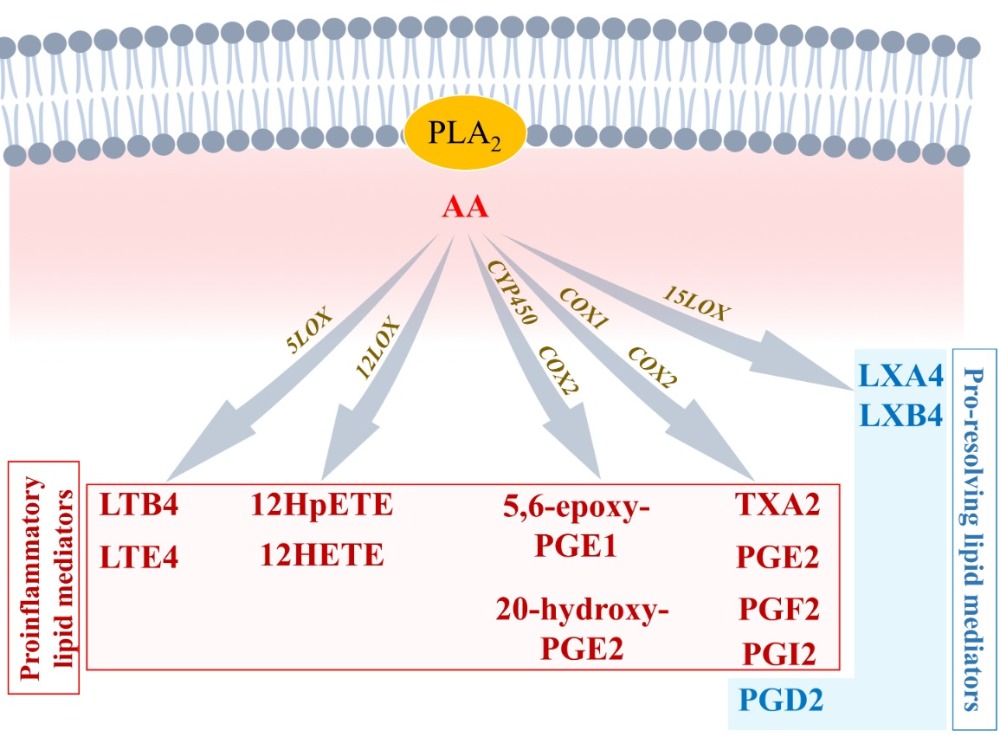
2.1.2. Arachidonic Acid Metabolism by COX1 and COX2
AA can directly interact with COX1 and COX2 to produce prostaglandin H2 (PGH2), an intermediate metabolite that is converted into bioactive proinflammatory lipid mediators such as thromboxane A2 (TXA2), prostaglandin A2 (PGA2), PGB2, PGE2, and PGI2 (Figure 2). These AA metabolites have been shown to be elevated in various cardiovascular conditions, including hypertension, atherosclerosis, vasculopathy, and myocardial infarction [29]. AA-derived lipids mediate vasoconstriction, increase vascular permeability, and stimulate expression of proinflammatory chemokines (complement component (C): C3b, C5a; chemokine C-X-C motif ligand 1 (CXCL1), CXCL2, CXCL8) and interleukins (IL1β, IL6, IL8, IL18, tumor necrosis factor alpha (TNFα)) to promote polymorphonuclear leukocytes (PMN) and proinflammatory-(M1)-macrophages chemotaxis and adhesion, by increasing expression of intercellular adhesion molecule 1 (ICAM1), vascular cell adhesion molecule 1 (VCAM1), and e-selectin (SELE), which act on endothelial cells to promote the adherence of neutrophils to the blood vessel wall [29][30]. These inflammatory biomarkers have been described to promote the development and progression of cardiovascular diseases including cardiac arrhythmias and AF [11][12][22].2.1.3. Arachidonic Acid Metabolism by 5-LOX
AA can also interact with 5-LOX to produce 5-Hydroperoxyeicosatetraenoic acid (5-HpETE), which promotes vasoconstriction. 5-HpETE can be metabolized either by leukotriene (LT) C-synthase to produce LTC4, LTD4, and LTE4, or by LTA-hydrolase to produce LTB4 via LTA4, which are all leukotrienes playing proinflammatory properties by amplifying PMN and M1 macrophages influx in the injured tissue [31] (Figure 2). HETEs have been shown to activate the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signaling to promote abnormal CM hypertrophy [32]. Evidence shows that AA-derived metabolites’ receptors are expressed on most cardiac cells including CM and FB [33]. In CM, inflammation signaling promotes NFκB activity and the assembling of the NACHT, LRR, and PYD domains containing protein 3 (NLRP3) inflammasome, leading to secretion of IL-1β and increased inflammatory status [34]. Patients with AF have shown increased expression of IL-1β and NLRP3 inflammasome components [35]. Normal initiation of inflammation must be followed by bio-molecularly orchestrated cellular processes aiming to terminate the inflammatory state and promote resolution [36]. In this purpose, lipid-mediator (LM) class switching is a key event that could be defined as a transition phase between the end of inflammation-initiation and the beginning of resolution [37].2.2. Lipid-Mediator Class Switching: Transition from Pro-Inflammatory to Pro-Resolution Signals
During the initiation phase of inflammation, neutrophils have an intense apoptotic and phagocytic activity [38]. This activates intracellular accumulation and extracellular secretion of 12/15-LOX in the damaged tissue. This accumulation of 12/15-LOX activates lipid-mediator-class switching from proinflammatory to pro-resolution mediators [39]. AA is then enzymatically metabolized by 12/15-LOX into lipoxins, including lipoxin (LX) A4 and LXB4 (Figure 2). LXA4 activates its transmembrane specific-receptor lipoxin A4 receptor or formyl peptide receptor 2 (ALX/FPR2) expressed on PMN and macrophages to limit further leukocyte trafficking, stimulate monocyte recruitment, promote anti-inflammatory (M2)-macrophage polarization, and activate phagocytosis and elimination of debris [39]. LXA4 has been shown to be significantly decreased in patients with chronic heart failure [40]. Recent studies have shown that LXA4 attenuates myocarditis by inhibiting NFκB and PI3K/Akt signaling pathways. 15-epi-LXA4 promotes initiation of resolution after myocardial infarction [41][42]. This activity of LXA4 suggests that, in arrhythmogenic conditions, anti-resolution signals promote the diminution of LXA4 production or/and LXA4-associated activity and signaling [42]. LXA4 could be an interesting candidate in the prevention of inflammation-induced substrate of arrhythmias, including AF.2.3. Resolution of Inflammation: SPMs-Mediated Efferocytosis and Homeostasis
Along with AA, other essential n3PUFAs are delivered with edema fluids at the site of injury. Among them, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) compete with AA to be enzymatically metabolized by either CYP450, 5LOX, 12LOX, 15LOX, or aspirin-acetylated COX2 [43]. Knowing that AA-derived metabolites are crucial in the initiation of normal inflammation, and that EPA and DHA products are important in resolution, it is understandable that optimal healthy conditions must promote a fair balance between AA, EPA, and DHA concentrations. Hence, in opposition to what has long been thought, it is not recommended to completely annihilate AA-derived metabolites (i.e., by using COX inhibitors) to guaranty homeostasis [44] (Figure 2 and Figure 3).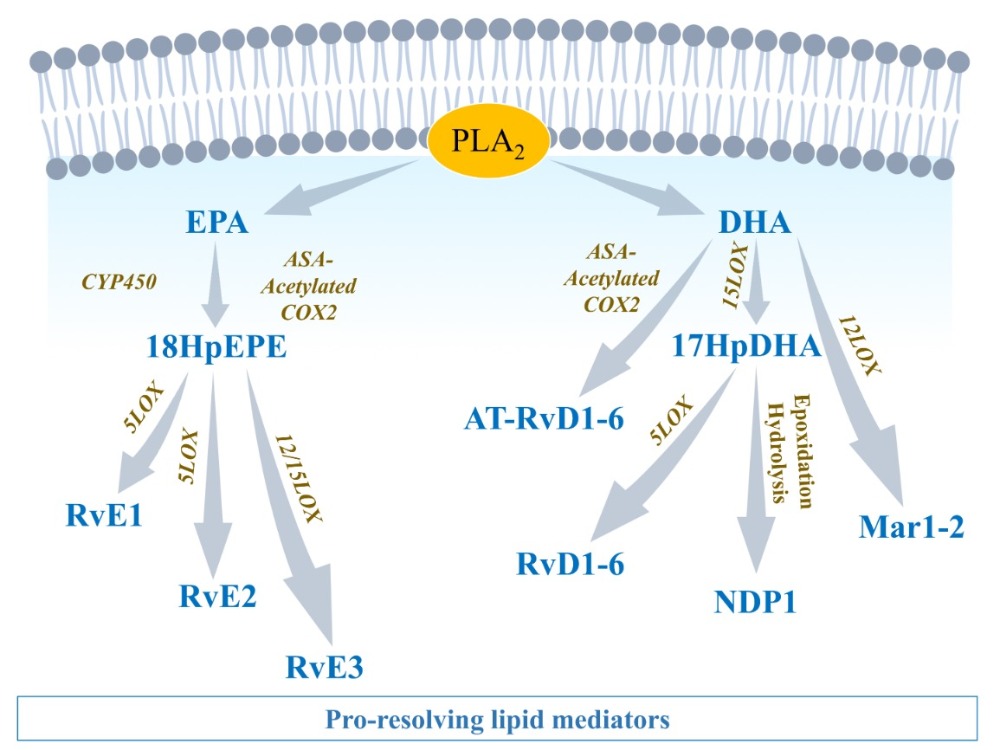
2.3.1. EPA-Derived Specialized Pro-Resolving Mediators
EPA is metabolized by CYP450 or aspirin-[ASA]-acetylated COX-2 into 18-HpEPE (18R-hydroperoxy-5Z, 8Z, 11Z, 14Z, 16E-eicosapentaenoic acid), which itself can interact with either 5LOX to produce E-series resolvins (Rvs), RvE1 and RvE2 or 15LOX to produce RvE3. E-series Rvs activate specific receptors such as chemokine-like receptor 1 (CMKLR1), also known as chemerin receptor 23 (ChemR23) (receptor of RvE1), or antagonize proinflammatory leukotriene receptors, such as leukotriene B4 receptor 1 (BLT1), expressed on PMN cell membrane, to stop the expression of chemoattractants, limit neutrophils adhesion/infiltration, and promote phagocytosis of apoptotic neutrophils and efferocytosis [45] (Figure 3).2.3.2. DHA-Derived Specialized Pro-Resolving Mediators
DHA can be metabolized by 12LOX to produce maresins (MaR1-2), or 15LOX to produce D-series resolvins (RvD1-6) and neuroprotectin D1 (NPD1) [46]. DHA interaction with aspirin-acetylated COX2 results in aspirin-triggered resolvins (AT-RvD1-6), which have been described to have similar properties as their classic homologs of the D-series Rvs [47] (Figure 3). D-series resolvins activate specific receptors such as ALX/FPR2 (receptor of RvD1), G-protein-coupled receptor 32 (GPR32: receptor of RvD1 and RvD3), and GPR18 (receptor of RvD2) that are expressed on vascular endothelial cells [37]. The activation of these signals promotes eNOS and P-ERK1/2 signaling, vascular permeability to non-phlogistic monocytes, cessation of PMN infiltration, macrophage polarization into M2-macrophages, M2-macrophages phagocytosis of cellular debris, and maintenance of homeostasis [37][45] (Figure 4).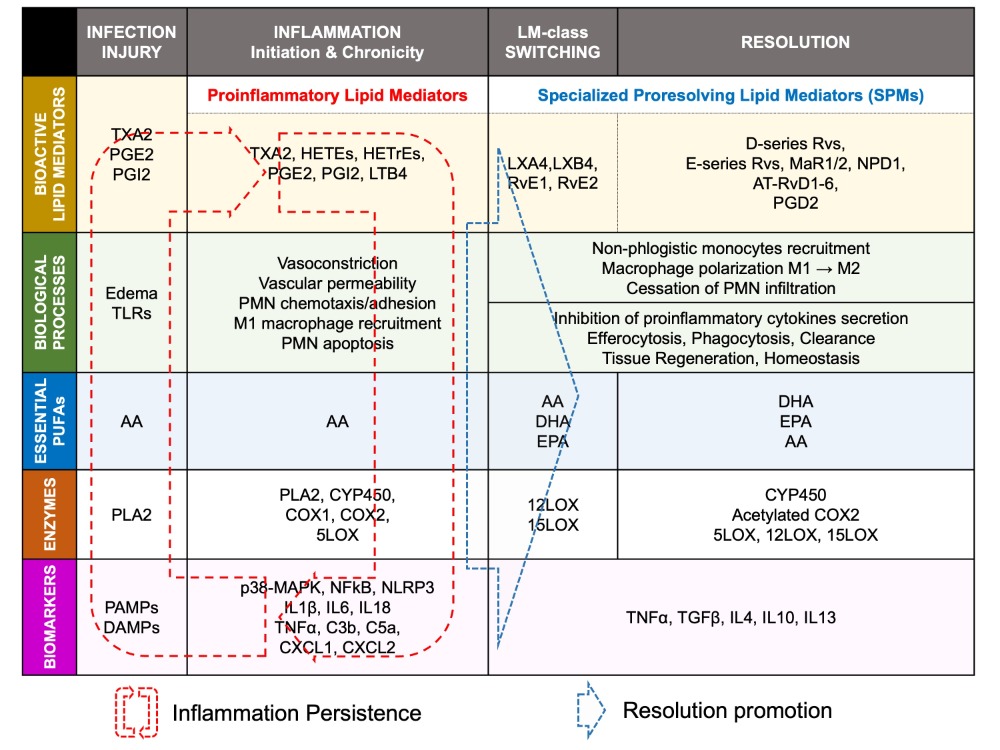
2.3.3. Arachidonic Acid-Derived Specialized Pro-Resolving Mediators
The metabolism of AA by COX does not only generate proinflammatory components. PGD2 has been shown to play an important role in resolution of inflammation [48]. PGD2 interacts with prostaglandin-D2-receptor 1 and 2 (DP1/2) expressed on T helper type (Th2) cells and dendritic cells that are involved in efferocytosis, phagocytosis, and clearance, to promote elimination of debris and pathogens, and induce complete resolution [48]. The interaction of AA with CYP450 can lead to production of epoxyeicosatrienoic acids (EETs) that are converted by soluble epoxide hydrolase (sEH) into dihydroxyeicosatrienoic acids (DiHETrEs). Although DiHETrEs are toxic, it has been shown that EETs mostly play a beneficial role by promoting vasorelaxation, and cardioprotective effects [49] (Figure 2 and Figure 4).2.4. ‘Failed Resolution Mechanisms’ in the Development of Chronic Inflammation and Heart Diseases
Lipid mediator (LM) production and signaling are fundamental in the regulation of the normal process of acute inflammation from its initiation to its resolution [46]. When the cardiac tissue undergoes a chronic inflammatory status, the crucial phase of LM class switching, which promotes the end of PMN infiltration and the activation of efferocytosis, may have failed to promote the shift of the cellular and lipidic accumulation from proinflammatory to pro-resolution mediators in the injured tissue [39]. Pathologic failure in the production of 12/15 LOX by immune cells including eosinophils, PMN, lymphocytes, and macrophages, leads to a lack of metabolization of AA into lipoxins (LXA4, LXB4) [38]. Lipoxins are essential to activate the cessation of neutrophil recruitment and the infiltration of non-phlogistic monocytes in the site of injury, which is the first step of resolution [38][39]. Moreover, lack of 12/15LOX prevents the production of D-series Rvs from DHA and RvE3 from EPA [38][50] (Figure 3). This may contribute to an annihilate resolution. Then, more proinflammatory LMs (Prostaglandins, leukotrienes) are produced from AA enzymatic interactions with the other enzymes available (COX2, CYP450, 5LOX) [50]. Abnormal accumulation of proinflammatory signaling promotes the prolongation of the initiation phase, characterized by the persistence of the external, cellular, and molecular signs of inflammation. This chronic inflammatory status leads to the development of fibrosis and loss of function [40][50]. If the local production of 12/15LOX is restored, or if the bioavailability of resolvins and lipoxins is increased (from diet or endogenous biosynthesis) at the site of injury, the tissue may enter the resolution phase via termination of proinflammatory signals, reduction of fibrosis, wound healing, and restoration of homeostasis [39][51][52] (Figure 1 and Figure 4). The detailed biomolecular characterization of inflammation–resolution remains partially understood in cardiac conditions. Moreover, each cardiac disease may display specific biomarkers involved in the incidence of the disorder. Although recent studies suggest a role of SPMs in ischemia-reperfusion [42][53] and pulmonary arterial hypertension-induced right atrial arrhythmogenic substrate [54], further fundamental research and clinical studies are required to assess whether resolution-promoting strategies and cytokine therapies could be an efficient approach to prevent and treat cardiac diseases, including hypertrophic cardiomyopathy, dilated cardiomyopathy, valvopathy, myocardial infarction, or arrhythmias.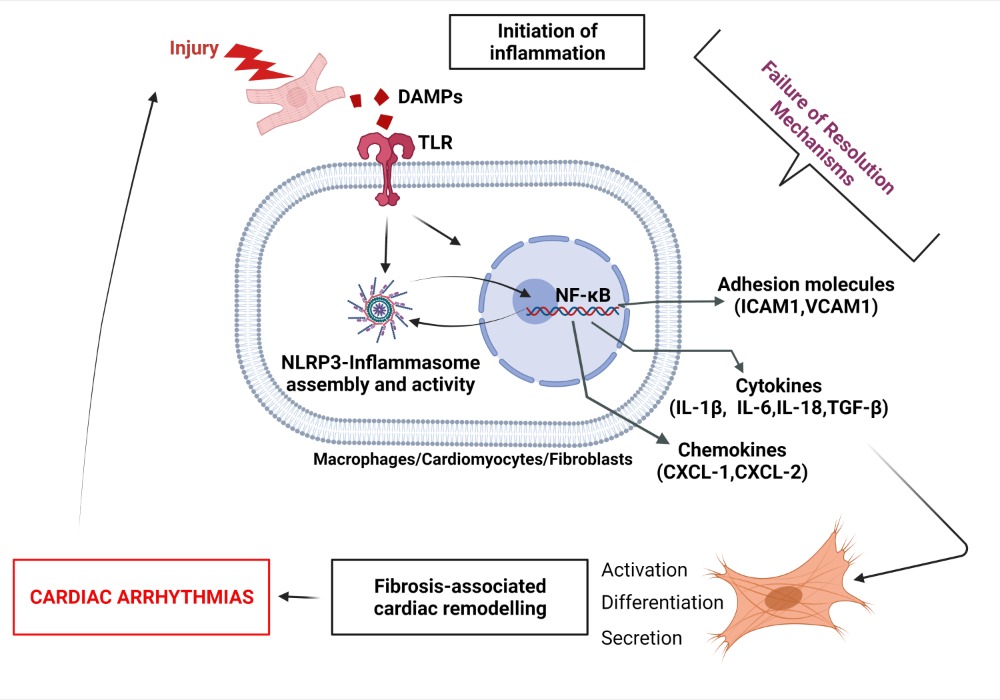
References
- Andrade, J.; Khairy, P.; Dobrev, D.; Nattel, S. The clinical profile and pathophysiology of atrial fibrillation: Relationships among clinical features, epidemiology, and mechanisms. Circ. Res. 2014, 114, 1453–1468.
- Hu, Y.-F.; Chen, Y.-J.; Lin, Y.-J.; Chen, S.-A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243.
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of Inflammation in Atrial Fibrillation Pathophysiology and Management. Circ. J. 2015, 79, 495–502.
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship Between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338.
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112.
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 160.
- Sun, Z.; Zhou, D.; Xie, X.; Wang, S.; Wang, Z.; Zhao, W.; Xu, H.; Zheng, L. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res. Cardiol. 2016, 111, 63.
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2013, 70, 74–82.
- Frangogiannis, N.G. The immune system and cardiac repair. Pharmacol. Res. 2008, 58, 88–111.
- Whitehead, A.J.; Engler, A.J. Regenerative cross talk between cardiac cells and macrophages. Am. J. Physiol. Circ. Physiol. 2021, 320, H2211–H2221.
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis—Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250.
- Scott, L., Jr.; Li, N.; Dobrev, D. Role of inflammatory signaling in atrial fibrillation. Int. J. Cardiol. 2019, 287, 195–200.
- Hiram, R. Cardiac cytokine therapy? Relevance of targeting inflammatory mediators to combat cardiac arrhythmogenic remodeling. IJC Heart Vasc. 2021, 37, 100918.
- Akdis, D.; Chen, K.; Saguner, A.M.; Stämpfli, S.F.; Chen, X.; Chen, L.; Rao, M.; Haegeli, L.M.; Tanner, F.C.; Brunckhorst, C.; et al. Clinical Characteristics of Patients with a Right Ventricular Thrombus in Arrhythmogenic Right Ventricular Cardiomyopathy. Thromb. Haemost. 2019, 119, 1373–1378.
- Keramida, K.; Lazaros, G.; Nihoyannopoulos, P. Right ventricular involvement in hypertrophic cardiomyopathy: Patterns and implications. Hell. J. Cardiol. 2018, 61, 3–8.
- Tadic, M.; Cuspidi, C.; Pencic, B.; Sljivic, A.; Ivanovic, B.; Neskovic, A.; Scepanovic, R.; Celic, V. High-normal blood pressure impacts the right heart mechanics: A three-dimensional echocardiography and two-dimensional speckle tracking imaging study. Blood Press. Monit. 2014, 19, 145–152.
- Waligóra, M.; Tyrka, A.; Miszalski-Jamka, T.; Urbańczyk-Zawadzka, M.; Podolec, P.; Kopeć, G. Right atrium enlargement predicts clinically significant supraventricular arrhythmia in patients with pulmonary arterial hypertension. Heart Lung 2018, 47, 237–242.
- Scheel, P.J., III; Murray, B.; Tichnell, C.; James, C.A.; Tandri, H.; Calkins, H.; Chelko, S.P.; Gilotra, N.A. Arrhythmogenic Right Ventricular Cardiomyopathy Presenting as Clinical Myocarditis in Women. Am. J. Cardiol. 2021, 145, 128–134.
- Sun, X.-Q.; Abbate, A.; Bogaard, H.-J. Role of cardiac inflammation in right ventricular failure. Cardiovasc. Res. 2017, 113, 1441–1452.
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285, Erratum in Nat. Rev. Cardiol. 2021, 18, 735.
- Everett, T.H., IV; Olgin, J.E. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 2007, 4 (Suppl. 3), S24–S27.
- Harada, M.; Nattel, S. Implications of Inflammation and Fibrosis in Atrial Fibrillation Pathophysiology. Card. Electrophysiol. Clin. 2021, 13, 25–35.
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A (2) regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6.
- Sofogianni, A.; Alkagiet, S.; Tziomalos, K. Lipoprotein-associated Phospholipase A2 and Coronary Heart Disease. Curr. Pharm. Des. 2018, 24, 291–296.
- Madjid, M.; Ali, M.; Willerson, J.T. Lipoprotein-associated phospholipase A2 as a novel risk marker for cardiovascular disease: A systematic review of the literature. Tex. Heart Inst. J. 2010, 37, 25–39.
- Tjoelker, L.W.; Wilder, C.; Eberhardt, C.; Stafforinit, D.M.; Dietsch, G.; Schimpf, B.; Hooper, S.; Le Trong, H.; Cousens, L.S.; Zimmerman, G.A.; et al. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature 1995, 374, 549–553.
- Kroetz, D.L.; Zeldin, D. Cytochrome P450 pathways of arachidonic acid metabolism. Curr. Opin. Lipidol. 2002, 13, 273–283.
- Rossi, A.G.; O’Flaherty, J.T. Bioactions of 5-hydroxyicosatetraenoate and its interaction with platelet-activating factor. Lipids 1991, 26, 1184–1188.
- Marnett, L.J.; Rowlinson, S.W.; Goodwin, D.; Kalgutkar, A.S.; Lanzo, C.A. Arachidonic Acid Oxygenation by COX-1 and COX-2. J. Biol. Chem. 1999, 274, 22903–22906.
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704.
- Rådmark, O.; Samuelsson, B. 5-Lipoxygenase: Mechanisms of regulation. J. Lipid Res. 2009, 50, S40–S45.
- Maayah, Z.H.; El-Kadi, A.O.S. 5-, 12- and 15-Hydroxyeicosatetraenoic acids induce cellular hypertrophy in the human ventricular cardiomyocyte, RL-14 cell line, through MAPK- and NF-κB-dependent mechanism. Arch. Toxicol. 2015, 90, 359–373.
- Zhou, Y.; Khan, H.; Xiao, J.; Cheang, W.S. Effects of Arachidonic Acid Metabolites on Cardiovascular Health and Disease. Int. J. Mol. Sci. 2021, 22, 12029.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242.
- Pirault, J.; Bäck, M. Lipoxin and Resolvin Receptors Transducing the Resolution of Inflammation in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 1273.
- Recchiuti, A.; Mattoscio, D.; Isopi, E. Roles, Actions, and Therapeutic Potential of Specialized Pro-resolving Lipid Mediators for the Treatment of Inflammation in Cystic Fibrosis. Front. Pharmacol. 2019, 10, 252.
- Fiore, S.; Brezinski, M.E.; Sheppard, K.-A.; Serhan, C.N. The Lipoxin Biosynthetic Circuit and their Actions with Human Neutrophils. Adv. Exp. Med. Biol. 1991, 314, 109–132.
- Levy, B.D.; Clish, C.; ASchmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619.
- Reina-Couto, M.; Carvalho, J.; Valente, M.J.; Vale, L.; Afonso, J.; Carvalho, F.; Bettencourt, P.; Sousa, T.; Albino-Teixeira, A. Impaired resolution of inflammation in human chronic heart failure. Eur. J. Clin. Investig. 2014, 44, 527–538.
- Shi, Y.; Pan, H.; Zhang, H.-Z.; Zhao, X.-Y.; Jin, J.; Wang, H.-Y. Lipoxin A4 mitigates experimental autoimmune myocarditis by regulating inflammatory response, NF-κB and PI3K/Akt signaling pathway in mice. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1850–1859.
- Kain, V.; Liu, F.; Kozlovskaya, V.; Ingle, K.A.; Bolisetty, S.; Agarwal, A.; Khedkar, S.; Prabhu, S.D.; Kharlampieva, E.; Halade, G.V. Resolution Agonist 15-epi-Lipoxin A4 Programs Early Activation of Resolving Phase in Post-Myocardial Infarction Healing. Sci. Rep. 2017, 7, 9999.
- Ishihara, T.; Yoshida, M.; Arita, M. Omega-3 fatty acid-derived mediators that control inflammation and tissue homeostasis. Int. Immunol. 2019, 31, 559–567.
- Cairns, J.A. The coxibs and traditional nonsteroidal anti-inflammatory drugs: A current perspective on cardiovascular risks. Can. J. Cardiol. 2007, 23, 125–131.
- El Kebir, D.; Gjorstrup, P.; Filep, J.G. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 14983–14988.
- Serhan, C.N.; Arita, M.; Hong, S.; Gotlinger, K. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: An overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat. 2004, 73, 155–172.
- Duvall, M.G.; Levy, B.D. DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur. J. Pharmacol. 2015, 785, 144–155.
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arter. Thromb. Vasc. Biol. 2011, 31, 986–1000.
- Oni-Orisan, A.; Alsaleh, N.; Lee, C.R.; Seubert, J.M. Epoxyeicosatrienoic acids and cardioprotection: The road to translation. J. Mol. Cell. Cardiol. 2014, 74, 199–208.
- Serhan, C.N.; Gupta, S.K.; Perretti, M.; Godson, C.; Brennan, E.; Li, Y.; Soehnlein, O.; Shimizu, T.; Werz, O.; Chiurchiù, V.; et al. The Atlas of Inflammation Resolution (AIR). Mol. Asp. Med. 2020, 74, 100894.
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832.
- Qu, X.; Zhang, X.; Yao, J.; Song, J.; Nikolic-Paterson, D.J.; Li, J. Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. J. Pathol. 2012, 228, 506–519.
- Kain, V.; Ingle, K.A.; Colas, R.A.; Dalli, J.; Prabhu, S.D.; Serhan, C.N.; Joshi, M.D.; Halade, G.V. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J. Mol. Cell. Cardiol. 2015, 84, 24–35.
- Hiram, R.; Xiong, F.; Naud, P.; Xiao, J.; Sirois, M.; Tanguay, J.-F.; Tardif, J.-C.; Nattel, S. The inflammation-resolution promoting molecule resolvin-D1 prevents atrial proarrhythmic remodelling in experimental right heart disease. Cardiovasc. Res. 2020, 117, 1776–1789.