Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Julian Christoffer Bachmann | -- | 2081 | 2022-05-25 10:07:19 | | | |
| 2 | Nora Tang | Meta information modification | 2081 | 2022-05-26 08:38:35 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bachmann, J.; Herum, K.; , .; Borghetti, G. Dual Roles of Fibrosis in HFpEF and CAD. Encyclopedia. Available online: https://encyclopedia.pub/entry/23337 (accessed on 24 July 2026).
Bachmann J, Herum K, , Borghetti G. Dual Roles of Fibrosis in HFpEF and CAD. Encyclopedia. Available at: https://encyclopedia.pub/entry/23337. Accessed July 24, 2026.
Bachmann, Julian, Kate Herum, , Giulia Borghetti. "Dual Roles of Fibrosis in HFpEF and CAD" Encyclopedia, https://encyclopedia.pub/entry/23337 (accessed July 24, 2026).
Bachmann, J., Herum, K., , ., & Borghetti, G. (2022, May 25). Dual Roles of Fibrosis in HFpEF and CAD. In Encyclopedia. https://encyclopedia.pub/entry/23337
Bachmann, Julian, et al. "Dual Roles of Fibrosis in HFpEF and CAD." Encyclopedia. Web. 25 May, 2022.
Copy Citation
Patients with heart failure with preserved ejection fraction (HFpEF) and atherosclerosis-driven coronary artery disease (CAD) will have ongoing fibrotic remodeling both in the myocardium and in atherosclerotic plaques. However, the functional consequences of fibrosis differ for each location. Thus, cardiac fibrosis leads to myocardial stiffening, thereby compromising cardiac function, while fibrotic remodeling stabilizes the atherosclerotic plaque, thereby reducing the risk of plaque rupture.
extracellular matrix
heart failure
atherosclerosis
fibrous cap
cardiac fibroblast
1. Dual Roles of Fibrotic Remodeling in Cardiovascular Disease
Fibrosis, the accumulation of extracellular matrix (ECM), occurs in most diseases of the heart. Nevertheless, no anti-fibrotic drugs targeting the heart exist to date. This may, in part, be due to the dual role of fibrosis, being beneficial or detrimental depending on the context. The replacement fibrosis that occurs in response to myocardial infarction (MI) can be lifesaving as the rapid production of ECM substitutes dying cardiomyocytes, thereby, to some extent, maintaining the mechanical integrity of the myocardial tissue. Although myocardial function will be compromised, and these patients are at risk of developing heart failure with reduced ejection fraction (HFrEF), fibrotic remodeling prevents the ventricle from rupturing, which would otherwise lead to sudden death [1]. Thus, fibrosis is crucial for providing mechanical strength after acute injury. In other more chronic contexts, fibrotic remodeling is detrimental to tissue function. During hypertrophic remodeling, i.e., in the pressure-overloaded heart, excessive fibrotic remodeling causes stiffening of the myocardium and severely compromises the diastolic function of the heart. This type of fibrosis is called “reactive fibrosis” and presents as interstitial fibrosis (deposited between cardiomyocytes) and perivascular (deposited around the vasculature) [2]. Reactive fibrosis is thought to be a main driver for development of heart failure with preserved ejection fraction (HFpEF).
HFpEF, which accounts for more than half of all heart failure cases and is rising progressively [3][4], is defined by reduced filling of the ventricle during diastole while the contraction in systole is preserved. Myocardial stiffening is the main cause of reduced ventricular filling. Although myocardial stiffening can be caused by the impaired relaxation of cardiomyocytes [5], there is a strong correlation between myocardial extracellular volume fraction with death and hospitalization of heart failure patients [6], indicating that cardiac fibrosis is the main driver of myocardial stiffening [7][8]. Furthermore, endothelial-independent coronary microvascular dysfunction (CMD) is prominent in HFpEF patients [9], indicating that perivascular fibrosis increases microvascular resistance in this patient sub-group. Thus, there is compelling evidence that reducing cardiac fibrosis would be beneficial for HFpEF patients.
It is important to understand the complicated and heterogenous nature of cardiovascular disease, where patients often present with multiple cardiovascular complications. Particularly HFpEF patients are a heterogenic group with one or more of the following conditions: obesity, diabetes, hypertension and advanced age. These conditions are also substantial risk factors for coronary artery disease (CAD). Indeed, a recent study by Rush et al. [9] demonstrated that 51% of the studied HFpEF patients also had obstructive epicardial CAD and that this condition was unrecognized prior to the study examination.
CAD is usually caused by atherosclerosis, the formation of a plaque inside the arterial wall. Fibrotic remodeling is crucial for the stabilization and healing of the atherosclerotic plaque [10] as the extracellular matrix is the main component of the fibrous cap that separates the highly thrombogenic necrotic core of the atherosclerotic plaque from the blood. The rupture of the plaque and the subsequent formation of thrombi obstruct the flow and perfusion to parts of the myocardium, thereby causing MI, the main cause of death worldwide [11]. Typically, prone-to-rupture plaques are characterized by the presence of a large lipid-rich necrotic core enclosed by a thin fibrous cap that is poor in collagen [12], suggesting the inhibition of fibrotic remodeling and collagen production may destabilize the atherosclerotic plaque.
The described dual roles of fibrotic remodeling in myocardial stiffening and atherosclerotic plaque stability add another layer of complexity regarding the development of future cardiac anti-fibrotic drugs in patients with HFpEF and CAD. To clarify the similarities and differences in the regulation of the fibrotic remodeling of the myocardium and coronary atherosclerotic plaques, here show the main cell types and signaling pathways that regulate ECM production and degradation in each of these cardiac locations (Figure 1).
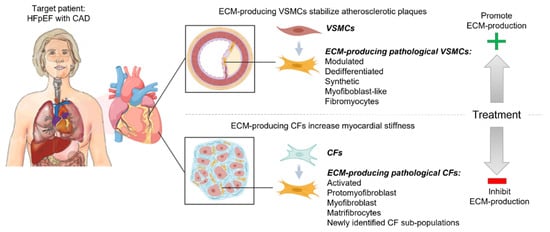
Figure 1. Dual roles of fibrotic remodeling in cardiovascular disease. Targeting ECM remodeling to reduce cardiac fibrosis in patients with Heart Failure with Preserved Ejection Fraction (HFpEF) and atherosclerosis-driven coronary artery disease (CAD) may entail a risk of destabilizing coronary atherosclerotic plaques, thereby causing myocardial infarction (MI). Cardiac fibroblasts (CFs) and vascular smooth muscle cells (VSMCs) regulate fibrotic remodeling of the myocardium and atherosclerotic plaque, respectively. They change phenotype in response to pathological stress and increase their production of ECM. These ECM-producing pathological CFs and VSMCs are given different names in the literature (as listed in the figure), depending on their phenotype, but may indicate similar subpopulations. These described cells are central for the induction of interstitial and perivascular fibrosis in HF patients and stabilization of the coronary atherosclerotic plaques in patients with CAD. The optimal treatment of patients with HFpEF and CAD should reduce cardiac fibrosis while maintaining, or promoting, a stable atherosclerotic plaque.
2. Cardiac Fibroblasts Regulate Fibrotic Remodeling of the Myocardium
Cardiac fibroblasts (CFs) are the main regulators of ECM production and maturation in the interstitial and perivascular areas of the heart. Under resting conditions, CFs maintain a healthy ECM by appropriately balancing production and degradation as well as regulating the composition and structure of the ECM [13]. Although a single unique molecular marker for CFs has not been identified, they are normally distinguished from other interstitial cells by the expression of collagen type I, the platelet-derived growth factor receptor alpha (PDGFRα), discoidin domain receptor tyrosine kinase 2 (DDR2) [14] and the transcription factor TCF21 [15][16][17]. In fact, TCF21 is required for the differentiation of epicardial cells into CFs [18], whereas the loss of TCF21 commits progenitors to coronary artery VSMC lineage [18][19]. Thus, TCF21 is decisive for the lineage fate of epicardial progenitor cells during development.
In response to pathological stress such as injury, chronic inflammation or changes in the mechanical environment, CFs become activated. Depending on the type of stress, fibroblast activation may involve one or more of the following functional effects: Increased proliferation, increased migration, excessive ECM production and acquisition of smooth muscle cell (SMC)-like features such as the ability to contract. Interestingly, TCF21 expression is lost in pathological conditions that induce fibrotic remodeling [20], suggesting that TCF21 may function as a master switch determining CF phenotype and pushing CFs toward an SMC-like phenotype.
The contractile property of activated fibroblasts was first described in 1971 by Gabbiani and colleagues, who introduced the cell ‘myofibroblast’ after an electron microscopy study of granulation tissue contractility. They induced contraction in strips of granulation tissue fibroblasts by agents known to contract the smooth muscle and observed functionally and morphologically characteristics close to that of SMCs [21]. Indeed, myofibroblasts are characterized by the presence of contractile α-smooth muscle actin (αSMA, encoded by the gene Acta2) filaments. However, recent data suggest that Acta2 is not required for the pro-fibrotic activity of myofibroblasts since the CF-specific deletion of Acta2 did not significantly affect contractility of myofibroblasts in vitro or cardiac repair and function following MI in vivo [22]. In agreement, early-stage myofibroblasts, named ‘proto-myofibroblasts’, are contractile despite a lack of αSMA [7]. In addition to their contractile function, myofibroblasts secrete excessive amounts of ECM proteins, such as collagens. Thus, myofibroblasts have long been known as key effector cells mediating ECM remodeling and fibrosis in most organs [7][8].
Myofibroblasts are suggested to derive from multiple cell types, including endothelial cells [23], bone marrow-derived cells and macrophages. However, compelling lineage-tracing studies using periostin (Postn) and Tcf21 to label myofibroblasts and resting CFs, respectively, identified resident CFs as the main source of myofibroblasts in mouse models of heart disease [20][24][25]. Perivascular Gli1-positive mesenchymal stem cell-like cells are also shown to contribute to the myofibroblast pool in the mouse model of heart failure [26] and were recently confirmed in human myocardial tissue after MI [27].
Recent single-cell transcriptomics studies revealed several new CF sub-populations in the healthy and diseased hearts of animal models [28][29][30] and humans [17][31][32]. Thus, in addition to the well-studied myofibroblast, it seems that CFs are more phenotypically dynamic than previously thought. E.g., two new fibroblast sub-populations, Cilp-positive and Thbs4-positive, were identified in angiotensin II (Ang II)-infused mouse hearts where Cilp-positive were the most fibrogenic cell type of the two. Both Cilp- and Thbs4-positive fibroblasts populations were not identified as myofibroblasts based on the lack of Acta2 expression [30][33], thus, questioning the traditional view that myofibroblasts are the main protagonists in cardiac fibrosis. Furthermore, a sophisticated lineage tracing study by the Molkentin lab described the dynamic differentiation path of CFs during the course of cardiac remodeling following MI [34]. Interestingly, myofibroblasts further differentiated into a new stable phenotypic state referred to as matrifibrocytes, which were also present in the mature scars of patients with MI. Matrifibrocytes were characterized by the expression of ECM genes typically expressed by chondrocytes and osteoblasts (e.g., Comp and Chad), suggesting an adaptive response to the highly stiff collagenous fibrotic environment. Along these lines, CFs were found to adopt an osteoblast cell-like fate during heart muscle calcification [35]. Thus, it is becoming increasingly clear that CFs adopt several phenotypes that change dynamically during the course of the disease.
Convincing human single-cell RNAseq (scRNAseq) of diseased human hearts, with sampling more than three patients and having a sufficient cell depth, is rare. A recent publication by Koenig et al. laid the foundation and will hopefully be followed by two major studies this year [27][36] (bioRxiv). Interestingly, RUNX1 and Gli1 are highlighted as important transcription factors (TF) for myofibroblast differentiation [27]. Together with the findings in mice, it points towards the conserved disease role of Gli1 in myofibroblast differentiation and adds RUNX1 as a driver in humans. On the contrary, TCF21 was not reported as a marker of activated CFs [27][36]; thus, TCF21 might be a unique driver in mouse myofibroblast differentiation or important for the early onset of disease. It will be interesting to see if CF sub-populations produce ECM with specific compositional profiles and whether these activities can be recapitulated in vitro.
3. Vascular Smooth Muscle Cells Regulate Fibrotic Remodeling in Coronary Atherosclerotic Plaques
In the healthy state, VSMCs reside in the medial layer of the arterial wall and are defined by their contractile phenotypic state, which is essential for the regulation of vascular tone and structural integrity. They have minor proliferative and ECM-producing activity, secreting only low amounts of ECM components such as elastin, collagen and proteoglycans to provide structural support and elasticity to the vessel wall [37].
Although healthy VSMCs display a highly differentiated contractile state, they retain remarkable plasticity and, in response to pathological stimuli, undergo phenotypic switching [38][39] that is characterized by the loss of contractile markers, including ACTA2, SM myosin heavy chain 11 (MYH11) and SM22α (also referred as transgelin, TAGLN), and increase in their ability to produce ECM, proliferate and migrate [40][41].
The emergence of lineage-tracing transgenic mice enabled cell-type-specific labeling and fate mapping of VSMCs in different disease models regardless of the presence or absence of contractile markers [42][43][44]. These studies revealed that in atherosclerotic lesions, VSMCs might adopt several phenotypic states, including an osteo-chondrogenic phenotype that is present in advanced atherosclerotic plaques with ongoing calcification and a fibroblast-like phenotype, characterized by the reduced expression of contractile genes (ACTA2) and increased expression of the small leucine-rich proteoglycans (SLRPs), lumican, decorin and biglycan [19][43][45]. To illustrate the opposite phenotypic trajectory to fibroblast-derived myofibroblasts, this VSMC-derived phenotype was named a ‘fibromyocyte’. Considering the common features in CF-derived myofibroblasts and VSMC-derived fibromyocytes, it is tempting to speculate that there exists a continuous phenotypic spectrum ranging from CFs to VSMCs, where CF- and VSMC phenotypes move toward each other during disease. Indeed, the expression of the matricellular genes POSTN and SPP1, which are known to be expressed by activated CFs [46][47], was identified as key drivers of VSMC phenotypic switch in human atherosclerosis [48]. However, it is unclear to what extent VSMC-derived “fibromyocytes” exhibit a similar transcriptional profile to CF-derived myofibroblasts [19][45].
Tcf21, the transcription factor known to orchestrate differentiation of epicardial progenitor cells to either coronary artery VSMC or CF lineages, is central for phenotypic changes of both cell types during disease. Thus, Tcf21 induces VSMC switching into “fibromyocytes” [19][45], while the loss of Tcf21 expression is associated with the activation and differentiation of CFs into myofibroblasts [34]. Interestingly, Tcf21 has also been causally linked to CAD, where its reduced VSMC expression increases the risk of cardiovascular diseases. In fact, Tcf21 SMC-specific knock-out in ApoE−/− mice inhibits VSMC phenotypic modulation and limits their presence in the fibrous cap, supporting a protective role of TCF21 in atherosclerosis [45].
References
- Gao, X.M.; White, D.A.; Dart, A.M.; Du, X.J. Post-infarct cardiac rupture: Recent insights on pathogenesis and therapeutic interventions. Pharm. Ther. 2012, 134, 156–179.
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574.
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 2006, 355, 251–259.
- Schiattarella, G.G.; Rodolico, D.; Hill, J.A. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 423–434.
- Van Heerebeek, L.; Franssen, C.P.; Hamdani, N.; Verheugt, F.W.; Somsen, G.A.; Paulus, W.J. Molecular and cellular basis for diastolic dysfunction. Curr. Heart Fail. Rep. 2012, 9, 293–302.
- Yang, E.Y.; Ghosn, M.G.; Khan, M.A.; Gramze, N.L.; Brunner, G.; Nabi, F.; Nambi, V.; Nagueh, S.F.; Nguyen, D.T.; Graviss, E.A.; et al. Myocardial Extracellular Volume Fraction Adds Prognostic Information Beyond Myocardial Replacement Fibrosis. Circ. Cardiovasc. Imaging 2019, 12, e009535.
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488.
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, jcs227900.
- Rush, C.J.; Berry, C.; Oldroyd, K.G.; Rocchiccioli, J.P.; Lindsay, M.M.; Touyz, R.M.; Murphy, C.L.; Ford, T.J.; Sidik, N.; McEntegart, M.B.; et al. Prevalence of Coronary Artery Disease and Coronary Microvascular Dysfunction in Patients With Heart Failure With Preserved Ejection Fraction. JAMA Cardiol. 2021, 6, 1130–1143.
- Vergallo, R.; Crea, F. Atherosclerotic Plaque Healing. N. Engl. J. Med. 2020, 383, 846–857.
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56.
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866.
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278.
- Cowling, R.T.; Yeo, S.J.; Kim, I.J.; Park, J.I.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Greenberg, B.H. Discoidin domain receptor 2 germline gene deletion leads to altered heart structure and function in the mouse. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H773–H781.
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409.
- Tallquist, M.D. Cardiac Fibroblast Diversity. Annu. Rev. Physiol. 2020, 82, 63–78.
- Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472.
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149.
- Nurnberg, S.T.; Cheng, K.; Raiesdana, A.; Kundu, R.; Miller, C.L.; Kim, J.B.; Arora, K.; Carcamo-Oribe, I.; Xiong, Y.; Tellakula, N.; et al. Coronary Artery Disease Associated Transcription Factor TCF21 Regulates Smooth Muscle Precursor Cells that Contribute to the Fibrous Cap. Genom. Data 2015, 5, 36–37.
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; SC, J.L.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 1–14.
- Majno, G.; Gabbiani, G.; Hirschel, B.J.; Ryan, G.B.; Statkov, P.R. Contraction of granulation tissue in vitro: Similarity to smooth muscle. Science 1971, 173, 548–550.
- Li, Y.; Li, C.; Liu, Q.; Wang, L.; Bao, A.X.; Jung, J.P.; Francis, J.; Molkentin, J.D.; Fu, X. Loss of Acta2 in cardiac fibroblasts does not affect myofibroblast differentiation or cardiac repair after myocardial infarction. BioRxiv 2021. version posted 21 May 2021.
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961.
- Moore-Morris, T.; Guimaraes-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934.
- Ivey, M.J.; Kuwabara, J.T.; Pai, J.T.; Moore, R.E.; Sun, Z.; Tallquist, M.D. Resident fibroblast expansion during cardiac growth and remodeling. J. Mol. Cell Cardiol. 2018, 114, 161–174.
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66.
- Kuppe, C.; Ramirez Flores, R.O.; Li, Z.; Hannani, M.; Tanevski, J.; Halder, M.; Cheng, M.; Ziegler, S.; Zhang, X.; Preisker, F.; et al. Spatial multi-omic map of human myocardial infarction. BioRxiv 2020. version posted 10 December 2020.
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; Nordon, R.E.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019, 8, e43882.
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610.
- McLellan, M.A.; Skelly, D.A.; Dona, M.S.I.; Squiers, G.T.; Farrugia, G.E.; Gaynor, T.L.; Cohen, C.D.; Pandey, R.; Diep, H.; Vinh, A.; et al. High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy. Circulation 2020, 142, 1448–1463.
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.C., Jr.; Akkad, A.D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020, 142, 466–482.
- Koenig, A.L.; Shchukina, I.; Amrute, J.; Andhey, P.S.; Zaitsev, K.; Lai, L.; Bajpai, G.; Bredemeyer, A.; Smith, G.; Jones, C.; et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat. Cardiovasc. Res. 2022, 1, 263–280.
- Krstevski, C.; Cohen, C.D.; Dona, M.S.I.; Pinto, A.R. New perspectives of the cardiac cellular landscape: Mapping cellular mediators of cardiac fibrosis using single-cell transcriptomics. Biochem. Soc. Trans. 2020, 48, 2483–2493.
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143.
- Pillai, I.C.L.; Li, S.; Romay, M.; Lam, L.; Lu, Y.; Huang, J.; Dillard, N.; Zemanova, M.; Rubbi, L.; Wang, Y.; et al. Cardiac Fibroblasts Adopt Osteogenic Fates and Can Be Targeted to Attenuate Pathological Heart Calcification. Cell Stem Cell 2017, 20, 218–232.e215.
- Linna-Kuosmanen, S.; Schmauch, E.; Galani, K.; Boix, C.A.; Hou, L.; Örd, T.; Toropainen, A.; Stolze, L.K.; Meibalan, E.; Mantero, J.C.; et al. Single-cell dissection of live human hearts in ischemic heart disease and heart failure reveals cell-type-specific driver genes and pathways. BioRxiv 2021. version posted 24 June 2021.
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801.
- Kurose, H. Cardiac Fibrosis and Fibroblasts. Cells 2021, 10, 1716.
- Herring, B.P.; Hoggatt, A.M.; Burlak, C.; Offermanns, S. Previously differentiated medial vascular smooth muscle cells contribute to neointima formation following vascular injury. Vasc. Cell 2014, 6, 1–14.
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc. Res. 2021, 117, 2326–2339.
- Basatemur, G.L.; Jorgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744.
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jorgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323.
- Jacobsen, K.; Lund, M.B.; Shim, J.; Gunnersen, S.; Fuchtbauer, E.M.; Kjolby, M.; Carramolino, L.; Bentzon, J.F. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight 2017, 2, e95890.
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637.
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289.
- Herum, K.M.; Romaine, A.; Wang, A.; Melleby, A.O.; Strand, M.E.; Pacheco, J.; Braathen, B.; Duner, P.; Tonnessen, T.; Lunde, I.G.; et al. Syndecan-4 Protects the Heart From the Profibrotic Effects of Thrombin-Cleaved Osteopontin. J. Am. Heart Assoc. 2020, 9, e013518.
- Snider, P.; Hinton, R.B.; Moreno-Rodriguez, R.A.; Wang, J.; Rogers, R.; Lindsley, A.; Li, F.; Ingram, D.A.; Menick, D.; Field, L.; et al. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ. Res. 2008, 102, 752–760.
- Alsaigh, T.; Evans, D.; Frankel, D.; Torkamani, A. Decoding the transcriptome of atherosclerotic plaque at single-cell resolution. BioRxiv 2020. version posted 4 March 2020.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
816
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
26 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No