 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yang Xiang | -- | 3401 | 2022-05-24 14:47:33 | | | |
| 2 | Peter Tang | Meta information modification | 3401 | 2022-05-25 03:27:19 | | |
Video Upload Options
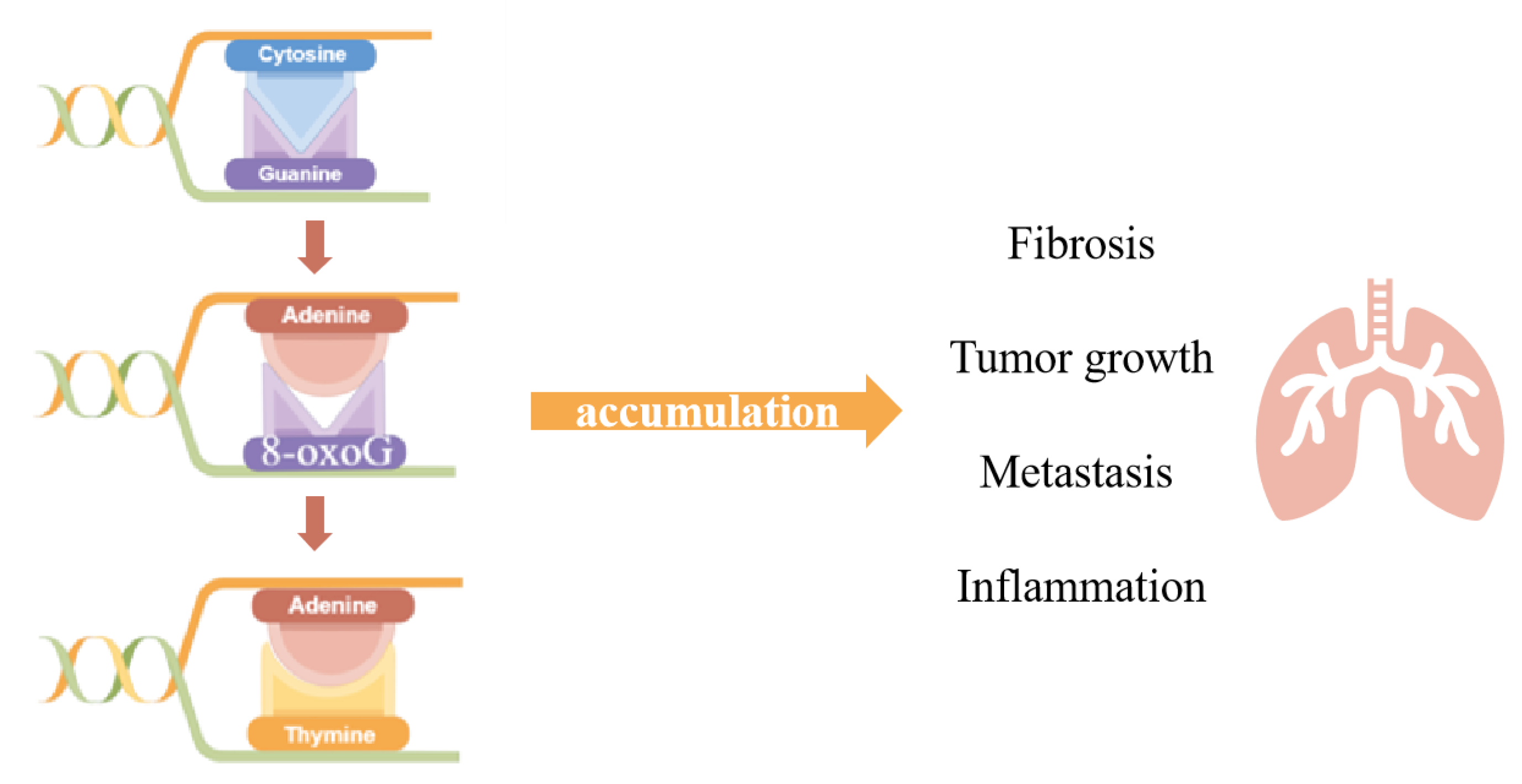
As the organ executing gas exchange and directly facing the external environment, the lungs are challenged continuously by various stimuli, causing the disequilibration of redox homeostasis and leading to pulmonary diseases. The breakdown of oxidants/antioxidants system happens when the overproduction of free radicals results in an excess over the limitation of cleaning capability, which could lead to the oxidative modification of macromolecules including nucleic acids. The most common type of oxidative base, 8-oxoG, is considered the marker of DNA oxidative damage. The appearance of 8-oxoG could lead to base mismatch and its accumulation might end up as tumorigenesis. The base 8-oxoG was corrected by base excision repair initiated by 8-oxoguanine DNA glycosylase-1 (OGG1), which recognizes 8-oxoG from the genome and excises it from the DNA double strand, generating an AP site for further processing. Aside from its function in DNA damage repairment, it has been reported that OGG1 takes part in the regulation of gene expression, derived from its DNA binding characteristic, and showed impacts on inflammation.
1. Introduction
2. The Role of OGG1 in DNA Oxidative Modification
3. The Roles of Base Excision Repair Enzyme OGG1 in Gene Expression
4. Roles of OGG1 in Pulmonary Inflammation and Disease
4.1. The Roles of OGG1 in Lung Cancer
4.2. The Roles of OGG1 in Innate Lung Immunity
4.3. The Roles of OGG1 in Airway Remodeling and Asthma
4.4. The Roles of OGG1 in Allergic Airway Inflammation
4.5. The Roles of OGG1 in Hyperoxia-Induced Lung Injury
References
- Sies, H.; Berndt, C.; Jones, D. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748.
- Wang, Y.; Yang, J.; Yi, J. Redox sensing by proteins: Oxidative modifications on cysteines and the consequent events. Antioxid. Redox Signal. 2012, 16, 649–657.
- Sies, H.; Jones, D. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Whitsett, J.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35.
- Hayes, J.; Dinkova-Kostova, A.; Tew, K. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197.
- Liao, Z.; Chua, D.; Tan, N. Reactive oxygen species: A volatile driver of field cancerization and metastasis. Mol. Cancer 2019, 18, 65.
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040.
- Tabner, B.; Turnbull, S.; El-Agnaf, O.; Allsop, D. Production of reactive oxygen species from aggregating proteins implicated in Alzheimer’s disease, Parkinson’s disease and other neurodegenerative diseases. Curr. Top. Med. Chem. 2001, 1, 507–517.
- Tieu, K.; Ischiropoulos, H.; Przedborski, S. Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life 2003, 55, 329–335.
- Hensley, K.; Butterfield, D.; Hall, N.; Cole, P.; Subramaniam, R.; Mark, R.; Mattson, M.; Markesbery, W.; Harris, M.; Aksenov, M. Reactive oxygen species as causal agents in the neurotoxicity of the Alzheimer’s disease-associated amyloid beta peptide. Ann. N. Y. Acad. Sci. 1996, 786, 120–134.
- Multhaup, G.; Ruppert, T.; Schlicksupp, A.; Hesse, L.; Beher, D.; Masters, C.; Beyreuther, K. Reactive oxygen species and Alzheimer’s disease. Biochem. Pharmacol. 1997, 54, 533–539.
- Dugan, L.; Quick, K. Reactive oxygen species and aging: Evolving questions. Sci. Aging Knowl. Environ. 2005, 2005, pe20.
- Lau, A.; Wang, Y.; Chiu, J. Reactive oxygen species: Current knowledge and applications in cancer research and therapeutic. J. Cell. Biochem. 2008, 104, 657–667.
- Renschler, M. The emerging role of reactive oxygen species in cancer therapy. Eur. J. Cancer 2004, 40, 1934–1940.
- Weinberg, F.; Chandel, N. Reactive oxygen species-dependent signaling regulates cancer. Cell. Mol. Life Sci. 2009, 66, 3663–3673.
- Atabek, M.; Vatansev, H.; Erkul, I. Oxidative stress in childhood obesity. J. Pediatr. Endocrinol. Metab. 2004, 17, 1063–1068.
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761.
- Yoshizumi, M.; Tsuchiya, K.; Tamaki, T. Signal transduction of reactive oxygen species and mitogen-activated protein kinases in cardiovascular disease. J. Med. Investig. 2001, 48, 11–24.
- Kinnula, V.; Fattman, C.; Tan, R.; Oury, T. Oxidative stress in pulmonary fibrosis: A possible role for redox modulatory therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 417–422.
- Di Virgilio, F. New pathways for reactive oxygen species generation in inflammation and potential novel pharmacological targets. Curr. Pharm. Des. 2004, 10, 1647–1652.
- Gelderman, K.; Hultqvist, M.; Olsson, L.; Bauer, K.; Pizzolla, A.; Olofsson, P.; Holmdahl, R. Rheumatoid arthritis: The role of reactive oxygen species in disease development and therapeutic strategies. Antioxid. Redox Signal. 2007, 9, 1541–1567.
- Gaston, B.; Drazen, J.; Loscalzo, J.; Stamler, J. The biology of nitrogen oxides in the airways. Am. J. Respir. Crit. Care Med. 1994, 149, 538–551.
- Shi, X.; Ding, M.; Chen, F.; Wang, L.; Rojanasakul, Y.; Vallyathan, V.; Castranova, V. Reactive oxygen species and molecular mechanism of silica-induced lung injury. J. Environ. Pathol. Toxicol. Oncol. 2001, 20 (Suppl. 1), 85–93.
- Rahman, I.; MacNee, W. Regulation of redox glutathione levels and gene transcription in lung inflammation: Therapeutic approaches. Free Radic. Biol. Med. 2000, 28, 1405–1420.
- Di Stefano, A.; Caramori, G.; Oates, T.; Capelli, A.; Lusuardi, M.; Gnemmi, I.; Ioli, F.; Chung, K.; Donner, C.; Barnes, P.; et al. Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. Eur. Respir. J. 2002, 20, 556–563.
- Friedrich, B.; Schmidt, R.; Reiss, I.; Günther, A.; Seeger, W.; Müller, M.; Thul, J.; Schranz, D.; Gortner, L. Changes in biochemical and biophysical surfactant properties with cardiopulmonary bypass in children. Crit. Care Med. 2003, 31, 284–290.
- Zhang, Y.; Rohde, L.; Wu, H. Involvement of nucleotide excision and mismatch repair mechanisms in double strand break repair. Curr. Genom. 2009, 10, 250–258.
- Jalal, S.; Earley, J.; Turchi, J. DNA repair: From genome maintenance to biomarker and therapeutic target. Clin. Cancer Res. 2011, 17, 6973–6984.
- Malewicz, M.; Perlmann, T. Function of transcription factors at DNA lesions in DNA repair. Exp. Cell Res. 2014, 329, 94–100.
- Brown, J.; O’Carrigan, B.; Jackson, S.; Yap, T. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37.
- Postel-Vinay, S.; Vanhecke, E.; Olaussen, K.; Lord, C.; Ashworth, A.; Soria, J. The potential of exploiting DNA-repair defects for optimizing lung cancer treatment. Nat. Rev. Clin. Oncol. 2012, 9, 144–155.
- Jackson, S.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078.
- Huertas, D.; Sendra, R.; Muñoz, P. Chromatin dynamics coupled to DNA repair. Epigenetics 2009, 4, 31–42.
- Barnes, D.; Lindahl, T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu. Rev. Genet. 2004, 38, 445–476.
- Margolin, Y.; Shafirovich, V.; Geacintov, N.; DeMott, M.; Dedon, P. DNA sequence context as a determinant of the quantity and chemistry of guanine oxidation produced by hydroxyl radicals and one-electron oxidants. J. Biol. Chem. 2008, 283, 35569–35578.
- Ming, X.; Matter, B.; Song, M.; Veliath, E.; Shanley, R.; Jones, R.; Tretyakova, N. Mapping structurally defined guanine oxidation products along DNA duplexes: Influence of local sequence context and endogenous cytosine methylation. J. Am. Chem. Soc. 2014, 136, 4223–4235.
- David, S.; O’Shea, V.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950.
- Hegde, M.; Hegde, P.; Arijit, D.; Boldogh, I.; Mitra, S. Human DNA Glycosylase NEIL1’s Interactions with Downstream Repair Proteins Is Critical for Efficient Repair of Oxidized DNA Base Damage and Enhanced Cell Survival. Biomolecules 2012, 2, 564–578.
- Hazra, T.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528.
- Liu, M.; Bandaru, V.; Bond, J.; Jaruga, P.; Zhao, X.; Christov, P.; Burrows, C.; Rizzo, C.; Dizdaroglu, M.; Wallace, S. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 4925–4930.
- Hegde, M.; Hazra, T.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47.
- Wiederhold, L.; Leppard, J.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.; Izumi, T.; Prasad, R.; Wilson, S.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220.
- Fromme, J.; Banerjee, A.; Verdine, G. DNA glycosylase recognition and catalysis. Curr. Opin. Struct. Biol. 2004, 14, 43–49.
- Huffman, J.; Sundheim, O.; Tainer, J. DNA base damage recognition and removal: New twists and grooves. Mutat. Res. 2005, 577, 55–76.
- Parikh, S.; Putnam, C.; Tainer, J. Lessons learned from structural results on uracil-DNA glycosylase. Mutat. Res. 2000, 460, 183–199.
- Bruner, S.; Norman, D.; Verdine, G. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 2000, 403, 859–866.
- Banerjee, A.; Yang, W.; Karplus, M.; Verdine, G. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 2005, 434, 612–618.
- Radom, C.; Banerjee, A.; Verdine, G. Structural characterization of human 8-oxoguanine DNA glycosylase variants bearing active site mutations. J. Biol. Chem. 2007, 282, 9182–9194.
- Amente, S.; Di Palo, G.; Scala, G.; Castrignanò, T.; Gorini, F.; Cocozza, S.; Moresano, A.; Pucci, P.; Ma, B.; Stepanov, I.; et al. Genome-wide mapping of 8-oxo-7,8-dihydro-2’-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res. 2019, 47, 221–236.
- Saxonov, S.; Berg, P.; Brutlag, D. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417.
- Chen, F.; Huang, D.; Chen, Y.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature 1998, 391, 410–413.
- Sakumi, K.; Tominaga, Y.; Furuichi, M.; Xu, P.; Tsuzuki, T.; Sekiguchi, M.; Nakabeppu, Y. Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res. 2003, 63, 902–905.
- Mabley, J.; Pacher, P.; Deb, A.; Wallace, R.; Elder, R.; Szabó, C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005, 19, 290–292.
- Li, G.; Yuan, K.; Yan, C.; Fox, J.; Gaid, M.; Breitwieser, W.; Bansal, A.; Zeng, H.; Gao, H.; Wu, M. 8-Oxoguanine-DNA glycosylase 1 deficiency modifies allergic airway inflammation by regulating STAT6 and IL-4 in cells and in mice. Free Radic. Biol. Med. 2012, 52, 392–401.
- Visnes, T.; Cázares-Körner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839.
- Pan, L.; Wang, H.; Luo, J.; Zeng, J.; Pi, J.; Liu, H.; Liu, C.; Ba, X.; Qu, X.; Xiang, Y.; et al. Epigenetic regulation of TIMP1 expression by 8-oxoguanine DNA glycosylase-1 binding to DNA:RNA hybrid. FASEB J. 2019, 33, 14159–14170.
- Donley, N.; Jaruga, P.; Coskun, E.; Dizdaroglu, M.; McCullough, A.; Lloyd, R. Small Molecule Inhibitors of 8-Oxoguanine DNA Glycosylase-1 (OGG1). ACS Chem. Biol. 2015, 10, 2334–2343.
- Chang, J.; Wrensch, M.; Hansen, H.; Sison, J.; Aldrich, M.; Quesenberry, C.; Seldin, M.; Kelsey, K.; Wiencke, J. Base excision repair genes and risk of lung cancer among San Francisco Bay Area Latinos and African-Americans. Carcinogenesis 2009, 30, 78–87.
- Hatt, L.; Loft, S.; Risom, L.; Møller, P.; Sørensen, M.; Raaschou-Nielsen, O.; Overvad, K.; Tjønneland, A.; Vogel, U. OGG1 expression and OGG1 Ser326Cys polymorphism and risk of lung cancer in a prospective study. Mutat. Res. 2008, 639, 45–54.
- Geng, P.; Yao, J.; Zhu, Y. hOGG1 Ser326Cys polymorphism and lung cancer susceptibility: A meta-analysis. Mol. Biol. Rep. 2014, 41, 2299–2306.
- Kiyohara, C.; Takayama, K.; Nakanishi, Y. Lung cancer risk and genetic polymorphisms in DNA repair pathways: A meta-analysis. J. Nucleic Acids 2010, 2010, 701760.
- Lan, Q.; Mumford, J.; Shen, M.; Demarini, D.; Bonner, M.; He, X.; Yeager, M.; Welch, R.; Chanock, S.; Tian, L.; et al. Oxidative damage-related genes AKR1C3 and OGG1 modulate risks for lung cancer due to exposure to PAH-rich coal combustion emissions. Carcinogenesis 2004, 25, 2177–2181.
- Kohno, T.; Kunitoh, H.; Mimaki, S.; Shiraishi, K.; Kuchiba, A.; Yamamoto, S.; Yokota, J. Contribution of the TP53, OGG1, CHRNA3, and HLA-DQA1 genes to the risk for lung squamous cell carcinoma. J. Thorac. Oncol. 2011, 6, 813–817.
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692.
- Baum, A.; Sachidanandam, R.; García-Sastre, A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 16303–16308.
- Choudhary, S.; Boldogh, I.; Brasier, A. Inside-Out Signaling Pathways from Nuclear Reactive Oxygen Species Control Pulmonary Innate Immunity. J. Innate Immun. 2016, 8, 143–155.
- Chen, H.; Chen, D.; Li, Y.; Yuan, W.; Fan, J.; Zhang, Z.; Han, F.; Jiang, X.; Chen, J.; Wang, D.; et al. Epigenetic silencing of TET1 mediated hydroxymethylation of base excision repair pathway during lung carcinogenesis. Environ. Pollut. 2021, 268, 115860.
- Wu, Z.; Turner, D.; Oliveira, D. IL-4 gene expression up-regulated by mercury in rat mast cells: A role of oxidant stress in IL-4 transcription. Int. Immunol. 2001, 13, 297–304.
- Zeyrek, D.; Cakmak, A.; Atas, A.; Kocyigit, A.; Erel, O. DNA damage in children with asthma bronchiale and its association with oxidative and antioxidative measurements. Pediatr. Allergy Immunol. 2009, 20, 370–376.
- Belanger, K.; Ameredes, B.; Boldogh, I.; Aguilera-Aguirre, L. The Potential Role of 8-Oxoguanine DNA Glycosylase-Driven DNA Base Excision Repair in Exercise-Induced Asthma. Mediat. Inflamm. 2016, 2016, 3762561.
- Ba, X.; Aguilera-Aguirre, L.; Sur, S.; Boldogh, I. 8-Oxoguanine DNA glycosylase-1-driven DNA base excision repair: Role in asthma pathogenesis. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 89–97.
- Bacsi, A.; Aguilera-Aguirre, L.; Szczesny, B.; Radak, Z.; Hazra, T.; Sur, S.; Ba, X.; Boldogh, I. Down-regulation of 8-oxoguanine DNA glycosylase 1 expression in the airway epithelium ameliorates allergic lung inflammation. DNA Repair 2013, 12, 18–26.
- Aguilera-Aguirre, L.; Hao, W.; Pan, L.; Li, X.; Saavedra-Molina, A.; Bacsi, A.; Radak, Z.; Sur, S.; Brasier, A.; Ba, X.; et al. Pollen-induced oxidative DNA damage response regulates miRNAs controlling allergic inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L1058–L1068.
- German, P.; Saenz, D.; Szaniszlo, P.; Aguilera-Aguirre, L.; Pan, L.; Hegde, M.; Bacsi, A.; Hajas, G.; Radak, Z.; Ba, X.; et al. 8-Oxoguanine DNA glycosylase1-driven DNA repair-A paradoxical role in lung aging. Mech. Ageing Dev. 2017, 161, 51–65.
- Kannan, S.; Pang, H.; Foster, D.C.; Rao, Z.; Wu, M. Human 8-oxoguanine DNA glycosylase increases resistance to hyperoxic cytotoxicity in lung epithelial cells and involvement with altered MAPK activity. Cell Death Differ. 2006, 13, 311–323.
- Wu, M.; He, Y.-H.; Kobune, M.; Xu, Y.; Kelley, M.R.; Martin, W.J. Protection of human lung cells against hyperoxia using the DNA base excision repair genes hOgg1 and Fpg. Am. J. Respir. Crit. Care Med. 2002, 166, 192–199.
- Ye, Y.; Lin, P.; Zhang, W.; Tan, S.; Zhou, X.; Li, R.; Pu, Q.; Koff, J.; Dhasarathy, A.; Ma, F.; et al. DNA Repair Interacts with Autophagy To Regulate Inflammatory Responses to Pulmonary Hyperoxia. J. Immunol. 2017, 198, 2844–2853.
- Thébaud, B.; Goss, K.; Laughon, M.; Whitsett, J.; Abman, S.; Steinhorn, R.; Aschner, J.; Davis, P.; McGrath-Morrow, S.; Soll, R.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78.
- Jiang, J.; Chou, H.; Chen, C. Cathelicidin attenuates hyperoxia-induced lung injury by inhibiting oxidative stress in newborn rats. Free Radic. Biol. Med. 2020, 150, 23–29.
- Jin, L.; Yang, H.; Fu, J.; Xue, X.; Yao, L.; Qiao, L. Association between oxidative DNA damage and the expression of 8-oxoguanine DNA glycosylase 1 in lung epithelial cells of neonatal rats exposed to hyperoxia. Mol. Med. Rep. 2015, 11, 4079–4086.

