 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sergio Oddi | -- | 2658 | 2022-05-24 11:57:01 | | | |
| 2 | Mauro Maccarrone | Meta information modification | 2658 | 2022-05-24 15:04:27 | | | | |
| 3 | Lindsay Dong | -21 word(s) | 2637 | 2022-05-25 10:29:20 | | |
Video Upload Options
Chronic inflammation in Alzheimer’s disease (AD) has been recently identified as a major contributor to disease pathogenesis. Once activated, microglial cells, which are brain-resident immune cells, exert several key actions, including phagocytosis, chemotaxis, and the release of pro- or anti-inflammatory mediators, which could have opposite effects on brain homeostasis, depending on the stage of disease and the particular phenotype of microglial cells. The endocannabinoids (eCBs) are pleiotropic bioactive lipids increasingly recognized for their essential roles in regulating microglial activity both under normal and AD-driven pathological conditions.
1. Introduction
2. Microglia
2.1. Microglial Functions and Phenotypes
2.2. Microglia and Alzheimer’s Disease
2.2.1. General Traits of Alzheimer’s Disease
Although a number of different environmental and genetic causes have been described in this pathology, the cause of the neurodegeneration has been historically linked to the aberrant accumulation of two hallmark protein aggregates, which represent pathognomonic signs of AD: (i) amyloid-β (Aβ) oligomers and polymers build up in the brain of affected people and constitute the typical plaques that are considered the main cause of neuronal loss and toxicity [19]; (ii) tau, a protein involved in stabilizing the microtubules and controlling axonal trafficking, undergoes pathological modifications that lead to its detachment from the microtubules and its aggregation to form tangles [20]. Even though amyloid plaques and tau tangles represent historical targets of investigation in this field, neuroinflammation has emerged in recent times as a central cause of neuronal loss in AD, with microglial cells considered to be a primary source in this process.
2.2.2. The Involvement of Microglia in Alzheimer’s Disease
3. The Endocannabinoid System
3.1. eCBs Synthesis and Degradation
3.2. eCBs Receptors and Molecular Pathways
4. Microglial Endocannabinoid System in Alzheimer’s Disease
4.1. Role of the ECS in Microglial Functionality
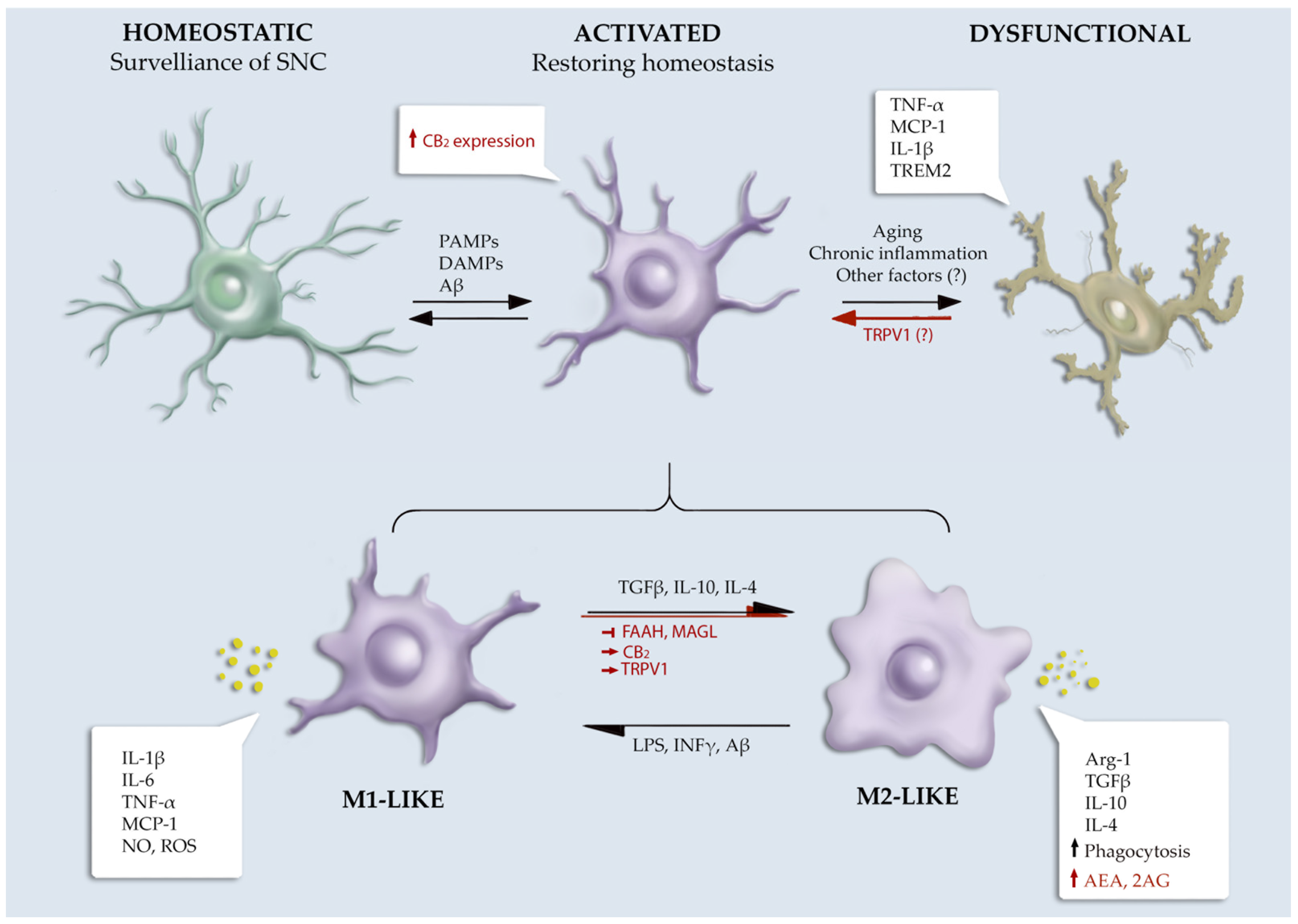
4.1.1. eCBs Receptors
4.1.2. eCBs Metabolic Enzymes
4.2. Alteration of ECS in Alzheimer’s Disease
Specific alterations in eCB signalling were observed in AD patients. In particular, based on the postmortem Braak staging method [67], CB1 was upregulated in the earliest stages [68][69], and downregulated in the advanced stages of AD [69][70][71]. However, other studies found conflicting results regarding the CB1 receptor expression, which remained unaffected in AD patients [57][72][73]. In human brains, CB2 receptor expression was found to be positively correlated with Aβ42 concentration, amyloid plaque burden, levels of hyperphosphorylated tau and neuritic tangles, consistent with the hypothesis that activated microglia could contribute to the inflammatory process of AD [57][70][71][74]. Other reports showed that the increase in the level of CB2 receptor was more pronounced in severe AD when compared with age-matched controls or moderate AD subjects [74]. CB2 mRNA expression in peripheral blood mononuclear cells (PBMCs) showed no differences between AD cases and controls [60]. Interestingly, in the brains of AD subjects, high levels of CB2 were found to be nitrosylated, an effect of the increase in peroxynitrite radicals attributable to microglia activation [70].
AD animal models are transgenic mice overexpressing mutant variants of human APP that provoke the accumulation of Aβ peptides and AD-like symptomatology [75].To accelerate/worsen the onset and the course of the amyloidosis, other models were developed by co-overexpressing other AD-related proteins, such as presenilin 1, apolipoprotein E (ApoE) and TREM2. All of these different types of AD-like models developed microgliosis and cognitive impairment, but with different time points of onset [76]. In some of these models, the expression and distribution of ECS elements were found with profound modifications compared to healthy mice.
4.3. Impact of Microglial Endocannabinoid Signalling in Alzheimer’s Disease
References
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398.
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56.
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and “wingmen”. Nat. Neurosci. 2015, 18, 800–806.
- Wong, W.T. Microglial aging in the healthy CNS: Phenotypes, drivers, and rejuvenation. Front. Cell. Neurosci. 2013, 7, 800–806.
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372.
- Streit, W.J.; Xue, Q.-S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142.
- Chiurchiu, V.; Battistini, L.; Maccarrone, M. Endocannabinoid signalling in innate and adaptive immunity. Immunology 2015, 144, 352–364.
- Stella, N. Endocannabinoid signaling in microglial cells. Neuropharmacology 2009, 56, 244–253.
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Bioactive lipids and chronic inflammation: Managing the fire within. Front. Immunol. 2018, 9, 38.
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2017, 18, 225–242.
- Mecha, M.; Feliú, A.; Carrillo-Salinas, F.J.; Rueda-Zubiaurre, A.; Ortega-Gutiérrez, S.; de Sola, R.G.; Guaza, C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav. Immun. 2015, 49, 233–245.
- Serhan, C.N.; Chiang, N.; Dalli, J. New Pro-Resolving n-3 Mediators Bridge Resolution of Infectious Inflammation to Tissue Regeneration. Mol. Aspects Med. 2018, 64, 1–17.
- Young, A.P.; Denovan-Wright, E.M. The Dynamic Role of Microglia and the Endocannabinoid System in Neuroinflammation. Front. Pharmacol. 2022, 12, 4069.
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172.
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16.
- Solé-Domènech, S.; Cruz, D.L.; Capetillo-Zarate, E.; Maxfield, F.R. The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res. Rev. 2016, 32, 89–103.
- Ji, Y.; Wang, X.; Kalicki, C.; Menta, B.W.; Baumgardner, M.; Koppel, S.J.; Weidling, I.W.; Perez-Ortiz, J.; Wilkins, H.M.; Swerdlow, R.H. Effects of Microglial Cytokines on Alzheimer’s Disease-Related Phenomena. J. Alzheimers. Dis. 2019, 67, 1021–1034.
- Guedes, J.R.; Lao, T.; Cardoso, A.L.; El Khoury, J. Roles of microglial and monocyte chemokines and their receptors in regulating Alzheimer’s disease-associated amyloid-β and tau pathologies. Front. Neurol. 2018, 9, 549.
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712.
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 22–35.
- Okello, A.; Edison, M.P.; Archer, M.H.; Turkheimer, M.F.; Kennedy, J.; Bullock, M.R.; Walker, M.Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial activation and amyloid deposition in mild cognitive impairment A PET study. Neurology 2009, 72, 56–62.
- Femminella, G.D.; Dani, M.; Wood, M.; Fan, Z.; Calsolaro, V.; Atkinson, R.; Edginton, T.; Hinz, R.; Brooks, D.J.; Edison, P. Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology 2019, 92, e1331–e1343.
- Hanzel, C.E.; Pichet-Binette, A.; Pimentel, L.S.B.; Iulita, M.F.; Allard, S.; Ducatenzeiler, A.; Do Carmo, S.; Cuello, A.C. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2249–2262.
- Philippens, I.H.; Ormel, P.R.; Baarends, G.; Johansson, M.; Remarque, E.J.; Doverskog, M. Acceleration of Amyloidosis by Inflammation in the Amyloid-Beta Marmoset Monkey Model of Alzheimer’s Disease. J. Alzheimers. Dis. 2017, 55, 101–113.
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475.
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2017, 101, 87–98.
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673.
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468.
- Maciuszek, M.; Cacace, A.; Brennan, E.; Godson, C.; Chapman, T.M. Recent advances in the design and development of formyl peptide receptor 2 (FPR2/ALX) agonists as pro-resolving agents with diverse therapeutic potential. Eur. J. Med. Chem. 2021, 213, 113167.
- Leuti, A.; Fazio, D.; Fava, M.; Piccoli, A.; Oddi, S.; Maccarrone, M. Bioactive lipids, inflammation and chronic diseases. Adv. Drug Deliv. Rev. 2020, 159, 133–169.
- Lutz, B.; Marsicano, G.; Maldonado, R.; Hillard, C.J. The endocannabinoid system in guarding against fear, anxiety and stress. Nat. Rev. Neurosci. 2015, 16, 705.
- Maccarrone, M.; Finazzi-Agró, A. The endocannabinoid system, anandamide and the regulation of mammalian cell apoptosis. Cell Death Differ. 2003, 10, 946–955.
- Binte Mustafiz, S.S.; Uyama, T.; Morito, K.; Takahashi, N.; Kawai, K.; Hussain, Z.; Tsuboi, K.; Araki, N.; Yamamoto, K.; Tanaka, T.; et al. Intracellular Ca2+-dependent formation of N-acyl-phosphatidylethanolamines by human cytosolic phospholipase A2ε. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2019, 1864, 158515.
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, Related Compounds and Their Metabolic Routes. Molecules 2014, 19, 17078.
- Liu, J.; Wang, L.; Harvey-White, J.; Osei-Hyiaman, D.; Razdan, R.; Gong, Q.; Chan, A.C.; Zhou, Z.; Huang, B.X.; Kim, H.Y.; et al. A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 2006, 103, 13345.
- Stella, N.; Schweitzer, P.; Plomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778.
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.J.; et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003, 163, 463–468.
- Hsu, K.L.; Tsuboi, K.; Adibekian, A.; Pugh, H.; Masuda, K.; Cravatt, B.F. DAGLβ inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat. Chem. Biol. 2012, 8, 999–1007.
- Nakane, S.; Oka, S.; Arai, S.; Waku, K.; Ishima, Y.; Tokumura, A.; Sugiura, T. 2-Arachidonoyl-sn-glycero-3-phosphate, an arachidonic acid-containing lysophosphatidic acid: Occurrence and rapid enzymatic conversion to 2-arachidonoyl-sn-glycerol, a cannabinoid receptor ligand, in rat brain. Arch. Biochem. Biophys. 2002, 402, 51–58.
- Higgs, H.N.; Glomset, J.A. Identification of a phosphatidic acid-preferring phospholipase A1 from bovine brain and testis. Proc. Natl. Acad. Sci. USA 1994, 91, 9574–9578.
- Murataeva, N.; Straiker, A.; Mackie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171, 1379–1391.
- Araujo, D.J.; Prakash, N.; Mechoulam, R.; Saijo, K.; Tang, Y.; Tjoa, K.; Guaza, C. The Endocannabinoid System as a Window into Microglial Biology and Its Relationship to Autism. Front. Cell. Neurosci. 2019, 13, 424.
- Pertwee, R.G. Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr. Med. Chem. 2010, 17, 1360–1381.
- Duffy, S.S.; Hayes, J.P.; Fiore, N.T.; Moalem-Taylor, G. The cannabinoid system and microglia in health and disease. Neuropharmacology 2021, 190, 108555.
- Klein, T.W. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat. Rev. Immunol. 2005, 5, 400–411.
- Ibsen, M.S.; Connor, M.; Glass, M. Cannabinoid CB1 and CB2 Receptor Signaling and Bias. Cannabis Cannabinoid Res. 2017, 2, 48.
- Connor, M.; Bagley, E.E.; Mitchell, V.A.; Ingram, S.L.; Christie, M.J.; Humphrey, P.P.A.; Vaughan, C.W. Cellular actions of somatostatin on rat periaqueductal grey neurons in vitro. Br. J. Pharmacol. 2004, 142, 1273.
- Ativie, F.; Komorowska, J.A.; Beins, E.; Albayram, Ö.; Zimmer, T.; Zimmer, A.; Tejera, D.; Heneka, M.; Bilkei-Gorzo, A. Cannabinoid 1 Receptor Signaling on Hippocampal GABAergic Neurons Influences Microglial Activity. Front. Mol. Neurosci. 2018, 11, 295.
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 2020, 11, 87.
- Scotter, E.L.; Abood, M.E.; Glass, M. The endocannabinoid system as a target for the treatment of neurodegenerative disease. Br. J. Pharmacol. 2010, 160, 480.
- Maccarrone, M.; Guzmán, M.; MacKie, K.; Doherty, P.; Harkany, T. Programming of neural cells by (endo)cannabinoids: From physiological rules to emerging therapies. Nat. Rev. Neurosci. 2014, 15, 786–801.
- Mecha, M.; Carrillo-Salinas, F.J.; Feliú, A.; Mestre, L.; Guaza, C. Microglia activation states and cannabinoid system: Therapeutic implications. Pharmacol. Ther. 2016, 166, 40–55.
- Cabral, G.A.; Harmon, K.N.; Carlisle, S.J. Cannabinoid-mediated inhibition of inducible nitric oxide production by rat microglial cells: Evidence for cb1 receptor participation. Adv. Exp. Med. Biol. 2001, 493, 207–214.
- Cutando, L.; Busquets-Garcia, A.; Puighermanal, E.; Gomis-González, M.; Delgado-García, J.M.; Gruart, A.; Maldonado, R.; Ozaita, A. Microglial activation underlies cerebellar deficits produced by repeated cannabis exposure. J. Clin. Investig. 2013, 123, 2816–2831.
- Lou, Z.Y.; Cheng, J.; Wang, X.R.; Zhao, Y.F.; Gan, J.; Zhou, G.Y.; Liu, Z.G.; Xiao, B.G. The inhibition of CB1 receptor accelerates the onset and development of EAE possibly by regulating microglia/macrophages polarization. J. Neuroimmunol. 2018, 317, 37–44.
- De Meij, J.; Alfanek, Z.; Morel, L.; Decoeur, F.; Leyrolle, Q.; Picard, K.; Carrier, M.; Aubert, A.; Séré, A.; Lucas, C.; et al. Microglial Cannabinoid Type 1 Receptor Regulates Brain Inflammation in a Sex-Specific Manner. Cannabis Cannabinoid Res. 2021, 6, 488–507.
- Benito, C.; Núñez, E.; Tolón, R.M.; Carrier, E.J.; Rábano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB 2 Receptors and Fatty Acid Amide Hydrolase Are Selectively Overexpressed in Neuritic Plaque-Associated Glia in Alzheimer’s Disease Brains. J. Neurosci. 2003, 23, 11136–11141.
- Gómez-Gálvez, Y.; Palomo-Garo, C.; Fernández-Ruiz, J.; García, C. Potential of the cannabinoid CB(2) receptor as a pharmacological target against inflammation in Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 200–208.
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6, 12.
- Mukhopadhyay, S.; Das, S.; Williams, E.A.; Moore, D.; Jones, J.D.; Zahm, D.S.; Ndengele, M.M.; Lechner, A.J.; Howlett, A.C. Lipopolysaccharide and cyclic AMP regulation of CB(2) cannabinoid receptor levels in rat brain and mouse RAW 264.7 macrophages. J. Neuroimmunol. 2006, 181, 82–92.
- Bisogno, T.; Oddi, S.; Piccoli, A.; Fazio, D.; Maccarrone, M. Type-2 cannabinoid receptors in neurodegeneration. Pharmacol. Res. 2016, 111, 721–730.
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 2010, 58, 1017–1030.
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic Cannabinoid Receptors Regulate Microglial Cell Migration. J. Neurosci. 2003, 23, 1398–1405.
- Muccioli, G.G.; Xu, C.; Odah, E.; Cudaback, E.; Cisneros, J.A.; Lambert, D.M.; Rodríguez, M.L.L.; Bajjalieh, S.; Stella, N. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J. Neurosci. 2007, 27, 2883–2889.
- Makara, J.K.; Mor, M.; Fegley, D.; Szabó, S.I.; Kathuria, S.; Astarita, G.; Duranti, A.; Tontini, A.; Tarzia, G.; Rivara, S.; et al. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat. Neurosci. 2005, 8, 1139–1141.
- Fernández-Suárez, D.; Celorrio, M.; Riezu-Boj, J.I.; Ugarte, A.; Pacheco, R.; González, H.; Oyarzabal, J.; Hillard, C.J.; Franco, R.; Aymerich, M.S. The monoacylglycerol lipase inhibitor JZL184 is neuroprotective and alters glial cell phenotype in the chronic MPTP mouse model. Neurobiol. Aging 2014, 35, 2603–2616.
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259.
- Farkas, S.; Nagy, K.; Palkovits, M.; Kovács, G.G.; Jia, Z.; Donohue, S.; Pike, V.; Halldin, C.; Máthé, D.; Harkany, T.; et al. SD-7015 reveals fine modalities of CB1 cannabinoid receptor density in the prefrontal cortex during progression of Alzheimer’s disease. Neurochem. Int. 2012, 60, 286–291.
- Manuel, I.; Lombardero, L.; LaFerla, F.M.; Giménez-Llort, L.; Rodríguez-Puertas, R. Activity of muscarinic, galanin and cannabinoid receptors in the prodromal and advanced stages in the triple transgenic mice model of Alzheimer’s disease. Neuroscience 2016, 329, 284–293.
- Ramírez, B.G.; Blázquez, C.; del Pulgar, T.G.; Guzmán, M.; de Ceballos, M.L. Prevention of Alzheimer’s Disease Pathology by Cannabinoids: Neuroprotection Mediated by Blockade of Microglial Activation. J. Neurosci. 2005, 25, 1904.
- Solas, M.; Francis, P.T.; Franco, R.; Ramirez, M.J. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 805–808.
- Westlake, T.M.; Howlett, A.C.; Bonner, T.I.; Matsuda, L.A.; Herkenham, M. Cannabinoid receptor binding and messenger RNA expression in human brain: An in vitro receptor autoradiography and in situ hybridization histochemistry study of normal aged and Alzheimer’s brains. Neuroscience 1994, 63, 637–652.
- Mulder, J.; Zilberter, M.; Pasquaré, S.J.; Alpár, A.; Schulte, G.; Ferreira, S.G.; Köfalvi, A.; Martín-Moreno, A.M.; Keimpema, E.; Tanila, H.; et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 2011, 134, 1041–1060.
- Halleskog, C.; Mulder, J.; Dahlström, J.; Mackie, K.; Hortobágyi, T.; Tanila, H.; Puli, L.K.; Färber, K.; Harkany, T.; Schulte, G. WNT Signaling in Activated Microglia Is Proinflammatory. Glia 2011, 59, 119–131.
- Hall, S.; Constantinescu, R.; Andreasson, U.; Surova, Y.; Bostrom, F.; Nilsson, C.; kan Widner, H.; Decraemer, H.; Nägga, K.; Minthon, L.; et al. Accuracy of a Panel of 5 Cerebrospinal Fluid Biomarkers in the Differential Diagnosis of Patients with Dementia and/or Parkinsonian Disorders. Arch. Neurol. 2012, 69, 1445–1452.
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89.
- Aso, E.; Palomer, E.; Juvés, S.; Maldonado, R.; Muñoz, F.J.; Ferrer, I. CB 1 Agonist ACEA Protects Neurons and Reduces the Cognitive Impairment of APP/PS1 Mice. J. Alzheimer’s Dis. 2012, 30, 439–459.
- Fakhfouri, G.; Ahmadiani, A.; Rahimian, R.; Grolla, A.A.; Moradi, F.; Haeri, A. WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-γ pathway. Neuropharmacology 2012, 63, 653–666.

