Chronic inflammation in Alzheimer’s disease (AD) has been recently identified as a major contributor to disease pathogenesis. Once activated, microglial cells, which are brain-resident immune cells, exert several key actions, including phagocytosis, chemotaxis, and the release of pro- or anti-inflammatory mediators, which could have opposite effects on brain homeostasis, depending on the stage of disease and the particular phenotype of microglial cells. The endocannabinoids (eCBs) are pleiotropic bioactive lipids increasingly recognized for their essential roles in regulating microglial activity both under normal and AD-driven pathological conditions.
1. Introduction
Compelling genetic and pathological evidence strongly supports the “amyloid cascade hypothesis” of AD, which states that beta-amyloid (Aβ), and in particular the least soluble 42-amino-acid-long Aβ isoform, is the causative agent in the pathogenesis of all forms of AD [
1,
2]. However, Aβ appears to be a necessary, yet insufficient, factor for AD development [
3]. Indeed, not all elderly patients who have significant amyloid plaque pathology develop the disease or show cognitive impairment, indicating that other ageing-promoting processes, by compromising the brain’s capacity to respond to Aβ peptides adequately, can determine the pathogenic evolution of amyloidosis [
3]. Recent evidence supports the idea that microglial dysregulation is indeed a crucial driver in the pathogenesis of AD [
4]. Altered/dystrophic microglial cells could contribute to aggravating and propagating an Aβ-pathologic cascade throughout the brain [
5,
6] by acquiring a “pro-AD” phenotype that consists of: (
i) the chronic release of pro-inflammatory cytokines and other inflammatory mediators, such as reactive oxygen and nitrogen species; (
ii) reduced phagocytosis and Aβ clearance; (
iii) reduced release of neurotrophic factors; (
iv) reduced release of pro-resolving factors; and (
v) reduced motility (chemotaxis).
The immune system can be modulated and regulated by humoral factors and metabolic products. Among these, eCBs are bioactive lipids that increase or decrease distinct immune functions when mobilized at the very beginning or shortly after first-line immune modulators [
7]. Notably, microglia express the array of receptors and metabolic enzymes (collectively termed “ECS”) that control the immune-related functions of eCBs [
8]. This system displays a wide distribution throughout the body and is involved in many adaptive responses to stressful internal and/or environmental insults. During the last few years, the brain ECS—by virtue of its capability of orchestrating neuromodulatory, anti-excitotoxic, anti-inflammatory/pro-resolutive, and anti-oxidative actions—has emerged as a key player in several neurodegenerative disorders, including AD [
9].
2. Microglia
Microglia, the resident immune defence in the brain, modulates the development, activity and plasticity of the central nervous system (CNS). To perform these complex functions, microglia adopt different activation states/phenotypes, depending on the microenvironment, which engage them in neuroinflammation, tissue repair, and even pro-resolutive inflammatory processes [
10,
11].
2.1. Microglial Functions and Phenotypes
Microglia represent highly versatile cells that play a pivotal role in the neurobiological, neuroinflammatory and neurophysiological homeostasis of the CNS in both health and disease. Often referred to as brain-resident macrophages—a definition that has been often criticized due to the ontogenetic and functional differences between these two cell types—microglia maintain neuroinflammatory homeostasis by interacting with other immune cells (e.g., T cells) and by releasing a vast array of pro- and anti-inflammatory cytokines and endogenous lipids, such as eCBs, arachidonic acid-derived autacoids and pro-resolving mediators [
12,
13,
14]; on the other hand, they also participate in pivotal brain functions, such as the elimination of dead neurons and cell debris [
14], synapse pruning and, to a minor extent, the regulation of the synaptic neurotransmitter tone.
In general, “resting” microglial cells exhibit a highly ramified appearance that gives them the ability to constantly survey the surrounding brain parenchyma, as they look for the presence of danger-, pathogen- or resolution-associated molecular patterns (DAMP, PAMP and RAMP, respectively) through their vast array of recognition systems represented by Toll-like receptors (TLRs), Nod-like receptors (NLRs), C-type lectin receptors (CLRs) and RIG-like receptors (RLRs), altogether referred to as pattern recognition receptors (PRRs) [
15,
16]. In particular, PRRs expressed on microglia surfaces recognize any molecular signal that indicates damage or cell stress, including ATP, nucleic acids, necrotic cells and cell debris and, relevant to this review, misfolded proteins such as Aβ species [
14,
15]. The recognition of any of these ligands leads to their internalization and the activation of microglia. Although in different neuroinflammatory and neurodegenerative paradigms, microglial cells have been reported to sport quite a vast array of morphologies, their pathological activation generally results in the transition towards an amoeboid phenotype that triggers potent neuroinflammatory reactions through the secretion of cytokines and chemokines [
17,
18]. Furthermore, depending on the nature of the recognized stimulus and/or the surrounding milieu to which they are exposed, microglia can assume different profiles, or phenotypes, the equilibrium of which contributes to the onset and outcome of neuroinflammatory and neurodegenerative mechanisms.
2.2. Microglia and Alzheimer’s Disease
2.2.1. General Traits of Alzheimer’s Disease
Although a number of different environmental and genetic causes have been described in this pathology, the cause of the neurodegeneration has been historically linked to the aberrant accumulation of two hallmark protein aggregates, which represent pathognomonic signs of AD: (i) amyloid-β (Aβ) oligomers and polymers build up in the brain of affected people and constitute the typical plaques that are considered the main cause of neuronal loss and toxicity [44]; (ii) tau, a protein involved in stabilizing the microtubules and controlling axonal trafficking, undergoes pathological modifications that lead to its detachment from the microtubules and its aggregation to form tangles [45]. Even though amyloid plaques and tau tangles represent historical targets of investigation in this field, neuroinflammation has emerged in recent times as a central cause of neuronal loss in AD, with microglial cells considered to be a primary source in this process.
2.2.2. The Involvement of Microglia in Alzheimer’s Disease
Microglia are profoundly involved in the correct functioning of the neuronal tissue, and overactivated or dysfunctional microglia are commonly featured in brain pathophysiology, cognitive decline, and AD, sometimes even preceding the neurological symptoms of the disease. In particular, microglia-induced neuroinflammation seems to represent a pivotal and necessary event in the development of the AD clinical phenotype. Indeed, microglial activation has been reported in patients with MCI- [
45,
46], even in the absence Aβ aggregates, and in animal models even before the formation of amyloid plaques [
46,
47,
48]. Further rationale supporting the role of early neuroinflammation in AD is provided by several recent studies. For example, the inheritance of pathological alleles for genes that are strongly involved in innate and microglial responses (e.g., TREM2, CD33 and complement proteins) represent a genetic risk for the development of AD (as reviewed in [
14]). Furthermore, microglia-mediated neuroinflammation has proven necessary, alongside aggregates, to induce the neuropathological features of the disease in animal models, and Aβ alone seems to be insufficient to induce symptoms [
49]; however, autoptic samples of asymptomatic patients with high Aβ brain loads also did not display activated microglial cells [
50]. The activation of microglia by Aβ contributed through the pyrin domain-containing 3 (NLRP3) inflammasome to enhance Aβ aggregation and to the tau progression [
51,
52].
Apparently, not only chronically-inflamed microglia is at the base of the clinical symptoms of AD, but the presence of Aβ seems to act as an enhancer, due to the fact microglial cells can recognize amyloid and tau aggregates as molecular profiles (i.e., PAMPs/DAMPs) through PRR-dependent signalling. This leads to the morphological changes of these cells and to the production of pro-inflammatory cytokines, such as IL-1β, IL-6, IL-8, TNF, as well as reactive oxygen species [
14]. This transition towards disease-associated microglia (DAM), which occurs with the progression of the disease, is characterized by a distinct transcriptomic fingerprint, which features the downregulation of homeostatic genes (e.g., purinergic receptors, Cx3cr1 and Tmem119) and the concomitant upregulation of AD-related genes such as ApoE, cathepsin D (Ctps), lipoprotein lipase (LPL), tyro protein tyrosine kinase binding protein (TyroBP) and Trem 2 [
40]. Of note, other receptors, such as formyl peptide receptor 2 (FPR2, also known as ALX), a G protein-coupled receptor that engages pro-resolving lipids, has been reported to bind Aβ [
53]. FPR2 displays strong ligand-biased signalling, and is expressed in microglia [
53], suggesting that AD pathology might also be related to interference between inflammation and resolution networks.
3. The Endocannabinoid System
3.1. eCBs Synthesis and Degradation
All cells in the body, including those of the immune system, produce AEA and 2-AG, the most studied eCBs. Rather than being pre-synthesized and stored in secretory vesicles, these bioactive lipids are made “on-demand” (i.e., when and where they are needed) by the receptor-stimulated cleavage of precursor membrane phosphoglycerides by several hydrolases.
AEA and 2-AG synthesis occurs through many alternative routes, which can also co-exist in the same cell and contribute to the production of eCBs in a time-, space- and activity-dependent manner [
57,
58]. AEA originates from a phospholipid precursor,
N-arachidonoylphosphatidyl ethanolamine (
NArPE), which is, in turn, formed from the
N-arachidonoylation of phosphatidylethanolamine via both Ca
2+-sensitive and Ca
2+-insensitive
N-acyltransferases (NATs and iNATs) [
59].
NArPE is then converted into AEA by several possible alternative pathways, the most direct of which is catalyzed by an
N-acylphosphatidylethanolamine-selective phosphodiesterase (NAPE-PLD) [
60]. In macrophages and other immune cells, another alternative biosynthetic pathway for AEA involves the PLC-catalyzed cleavage of
NArPE to yield phospho-AEA, which is subsequently dephosphorylated by protein tyrosine phosphatase non-receptor type 22 (PTPN22), a member of PEST family of protein tyrosine phosphatases [
61,
62].
Through the hydrolysis of different arachidonoyl-containing membrane lipids, 2-AG synthesis can potentially occur. The best-studied synthetic route for 2-AG is its synthesis from
sn-2-arachidonic-containing diacylglycerols (DAGs) [
63] by one of two DAG lipases (DAGL) isoforms, DAGLα and DAGLβ [
64]; the latter isoform is expressed more in macrophages, and although its relative brain expression is sparse, it is highly expressed in microglia [
65]. Alternatively, 2-AG can also be synthesized by the dephosphorylation of
sn-2 arachidonoyl-lysophosphatidic acid (LPA) [
66] via 2-LPA phosphatase; or by the sequential action of PLA1 and a lysophospholipase C (lyso-PLC) [
67] in
sn-2 arachidonate-containing phosphatidylinositol (PI) and its derivative 2-arachidonoyl-lysoPI, respectively [
68,
69].
3.2. eCBs Receptors and Molecular Pathways
CB
1 and CB
2 receptors are members of the superfamily A of the heptahelical transmembrane-spanning G protein-coupled receptors (GPCRs) coupled to heterotrimeric Gi/o proteins [
76,
77]. These receptors are expressed to various extents in immune cells, with CB
2 being predominant under physiological conditions and upon acute and chronic inflammation [
78]. Although CB
1 is primarily expressed in specific neuronal populations, with neuromodulatory activity, it also seems to play a role in regulating immune responses and inflammatory pathways [
79,
80]. In this context, Ativie and colleagues demonstrated that neuronal CB
1 may indirectly regulate microglial activity, possibly by influencing the crosstalk between neurons and microglia [
81].
The binding of eCBs to CB receptors affects several cellular pathways, such as the inhibition of adenylate cyclase and then of protein kinase A (PKA); the regulation of ionic currents (inhibition of voltage-gated L, N and P/Q-type Ca
2+ channels, activation of K
+ channels); the activation of focal adhesion kinases, such as MAPKs (p38, ERK1/2, JNK), PI3K/Akt and cytosolic phospholipase A2; and the activation (CB
1) or inhibition (CB
2) of iNOS. All of these pathways are involved in fundamental microglia functions [
57,
82,
83,
84].
4. Microglial Endocannabinoid System in Alzheimer’s Disease
4.1. Role of the ECS in Microglial Functionality
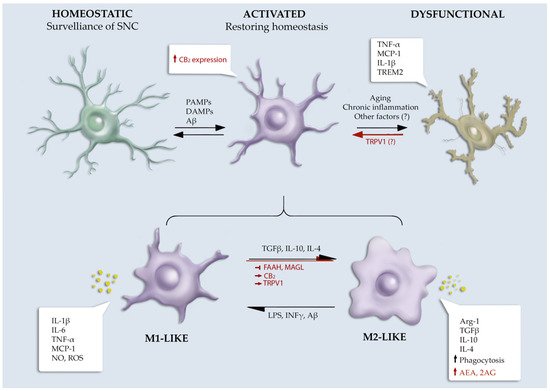
The ECS has a pivotal role in brain inflammation by regulating microglial biology in terms of proliferation, migration, phagocytosis, and the production of pro- and anti-inflammatory mediators [
8] (
Figure 1).
Figure 1. Schematic overview of ECS role in microglial phenotype. Symbols used: ↑, increased; ↓, decreased; →, activation; ⟞, inhibition.
4.1.1. eCBs Receptors
Although CB
1 was initially considered as a neuron-specific cannabinoid receptor, emerging evidence is revealing that this receptor is also constitutively expressed, even if at low levels, in microglia [
90,
91]. Indeed, a recent work with conditional knockout mice documented that hippocampal CB
1−/− microglia show a decreased expression of TNF-α compared to wild-type mice upon stimulation with LPS [
92]. However, to date, except for some rare works, the specific role of CB
1 in microglial cellular physiology has not been deeply explored [
92,
93,
94].
CB
2 immunoreactivity was primarily associated with astrocytes and microglial cells in the healthy brain. The expression of CB
2 is significantly upregulated in these cells following brain trauma or under other pathological conditions, including AD [
95], Parkinson’s disease [
96], and multiple sclerosis [
97]. Similar findings were observed in several mice models of neurodegenerative conditions [
98,
99]. The genetic ablation of CB
2 showed microglia with a reduced phagocytic capacity and relevant morphology alterations during the switching in M2 phenotype compared to wild-type cells. In particular, M2a microglia from CB
2−/− mice lost well-defined and multiple lamellipodia and took a more elongated shape. In addition, Arg-1 expression was diminished in CB
2−/− both under basal conditions and following M2a stimulation, suggesting a role for CB
2 in anti-inflammatory switching [
90].
4.1.2. eCBs Metabolic Enzymes
Microglia express a full assortment of synthetic and catabolic enzymes for eCBs and can therefore metabolize both 2-AG and AEA [
8,
109]. Effectively, microglia produce in vitro 20 times more eCBs than neurons and other glial cells and are likely to be the primary cellular source of these bioactive lipids under neuroinflammatory conditions [
110]. Moreover, eCBs metabolic enzymes seem to be regulated by microglial activation states [
110,
111]. In particular, stimulating the switch of microglia to either M2a or M2c states led to an upregulation of the biosynthetic enzymes (i.e., DAGLα in M2a; NAPE-PLD in M2c), accompanied by a downregulation of the respective degrading enzymes, thus resulting in the elevated production of eCBs [
90].
The pharmacological inhibition of MAGL was shown to raise 2-AG level in the brain [
112] and exerted beneficial immunomodulatory functions in neuroinflammatory conditions, attributable, at least in part, to the regulation of microglial functions. For example, in a PD mouse model, JZL184 (a selective MAGL inhibitor) prevented motor impairment and induced an increase in microglial cell number and ramification in the striatum, suggesting that MAGL activity impacted both microglial proliferation and phenotypes [
113].
4.2. Alteration of ECS in Alzheimer’s Disease
Specific alterations in eCB signalling were observed in AD patients. In particular, based on the postmortem Braak staging method [130], CB1 was upregulated in the earliest stages [131,132], and downregulated in the advanced stages of AD [102,132,133]. However, other studies found conflicting results regarding the CB1 receptor expression, which remained unaffected in AD patients [95,134,135]. In human brains, CB2 receptor expression was found to be positively correlated with Aβ42 concentration, amyloid plaque burden, levels of hyperphosphorylated tau and neuritic tangles, consistent with the hypothesis that activated microglia could contribute to the inflammatory process of AD [95,102,133,136]. Other reports showed that the increase in the level of CB2 receptor was more pronounced in severe AD when compared with age-matched controls or moderate AD subjects [136]. CB2 mRNA expression in peripheral blood mononuclear cells (PBMCs) showed no differences between AD cases and controls [98]. Interestingly, in the brains of AD subjects, high levels of CB2 were found to be nitrosylated, an effect of the increase in peroxynitrite radicals attributable to microglia activation [102].
AD animal models are transgenic mice overexpressing mutant variants of human APP that provoke the accumulation of Aβ peptides and AD-like symptomatology [143].To accelerate/worsen the onset and the course of the amyloidosis, other models were developed by co-overexpressing other AD-related proteins, such as presenilin 1, apolipoprotein E (ApoE) and TREM2. All of these different types of AD-like models developed microgliosis and cognitive impairment, but with different time points of onset [144]. In some of these models, the expression and distribution of ECS elements were found with profound modifications compared to healthy mice.
4.4. Impact of Microglial Endocannabinoid Signalling in Alzheimer’s Disease
Important information on the impact of the microglial ECS in AD has been obtained from preclinical studies performed on AD-like mice. CB
1 chronic activation by ACEA in APPSwe/PS1ΔE9 was effective in restoring cognitive dysfunction [
152]. Unfortunately, this study neglected to address the impact of CB
1 stimulation on microglia-driven neuroinflammation. Similarly, in a rat model where microglial activation and memory impairments were induced by Aβ injection, the activation of CB
1 by WIN55,212-2 (a non-selective CB
1/2 agonist) improved the cognitive deficit. Moreover, WIN55,212-2 prevented microglial activation in the cortex of Aβ-treated rats [
102]. In the same model, another group showed that WIN55,212-2, acting through CB
1 and CB
2 receptors, significantly improved memory functions, decreasing the expression of some neuroinflammatory markers, such as TNF-α, activated caspase-3, and nuclear NFκB [
153]. Additionally, the latter study did not address whether these effects could be ascribed to the cannabinoid-dependent modulation of microglial properties.
This entry is adapted from the peer-reviewed paper 10.3390/cells11071237