 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Els Verhoeyen | -- | 3826 | 2022-05-24 10:30:03 | | | |
| 2 | Beatrix Zheng | Meta information modification | 3826 | 2022-05-25 03:10:00 | | | | |
| 3 | Beatrix Zheng | -4 word(s) | 3822 | 2022-05-25 03:10:48 | | |
Video Upload Options
Escaping the immune system is the main characteristic of a tumor cell. In order to limit the anarchic proliferation of cancer cells, chemotherapies are often used in tumors that have no specific treatment yet, such as AITL. These therapies are proposed to limit the proliferation of dividing cells, i.e., healthy cells as well as tumor cells. This explains the poor benefits of such treatment and the major risk of relapse or even the absence of signs of remission. Importantly, targeting a specific cancer cell type without inducing an effect on healthy cells requires an in-depth knowledge of its phenotype and its surface markers, which can serve as an asset for the development of specific therapies, in particular immunotherapies. If some healthy cells are affected, it is called on-target off-target effects because certain cancer epitopes are partially expressed on non-malignant cells. These therapies include, for example, monoclonal antibodies and chimeric antigen receptors.
1. Introduction
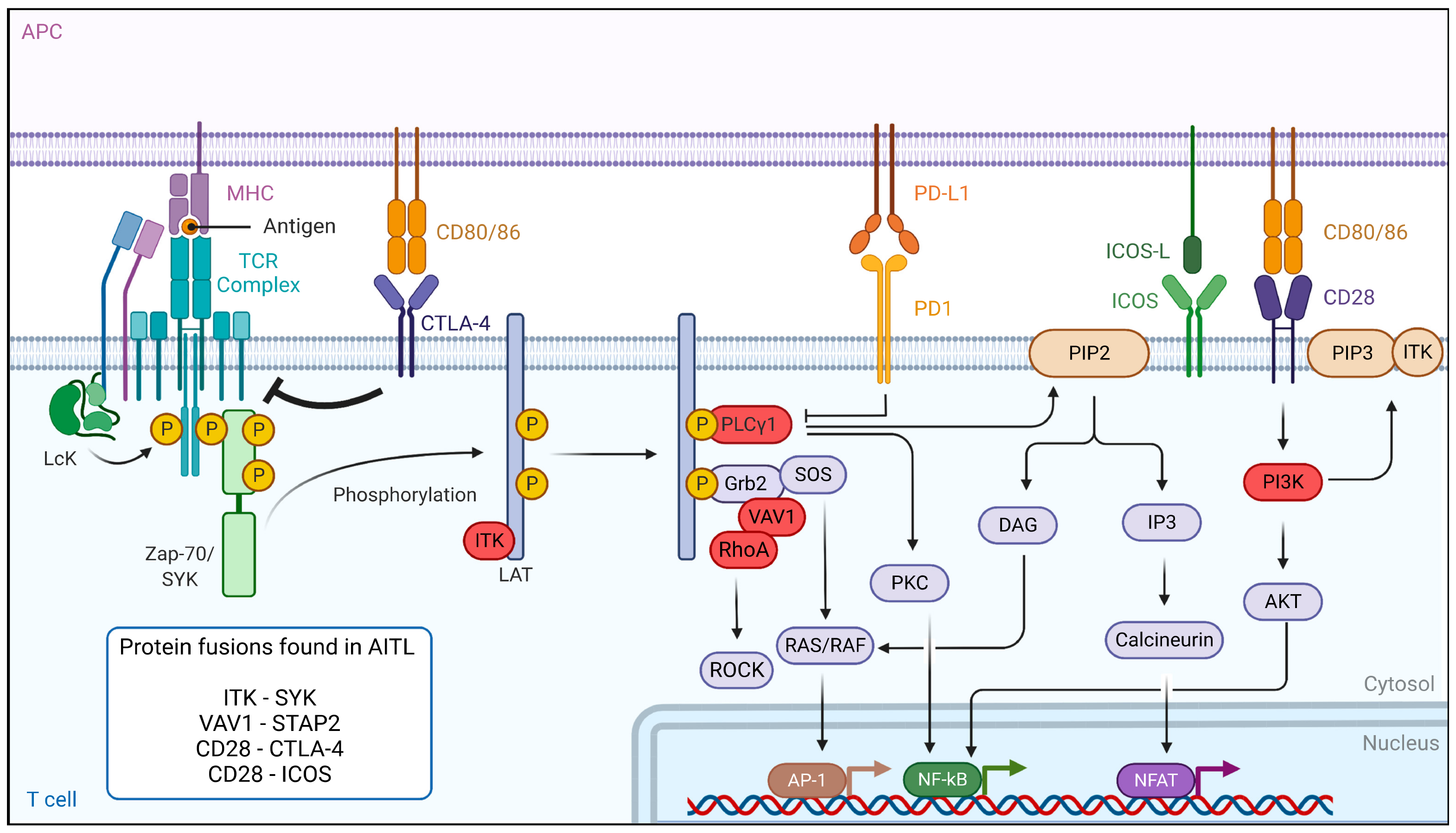
2. Dysregulation of the TCR Signaling Pathway in AITL Reveals New Treatment Options
3. Immunotherapeutic Approaches for AITL
3.1. Monoclonal-Antibody-Based Immunotherapies for AITL Treatment
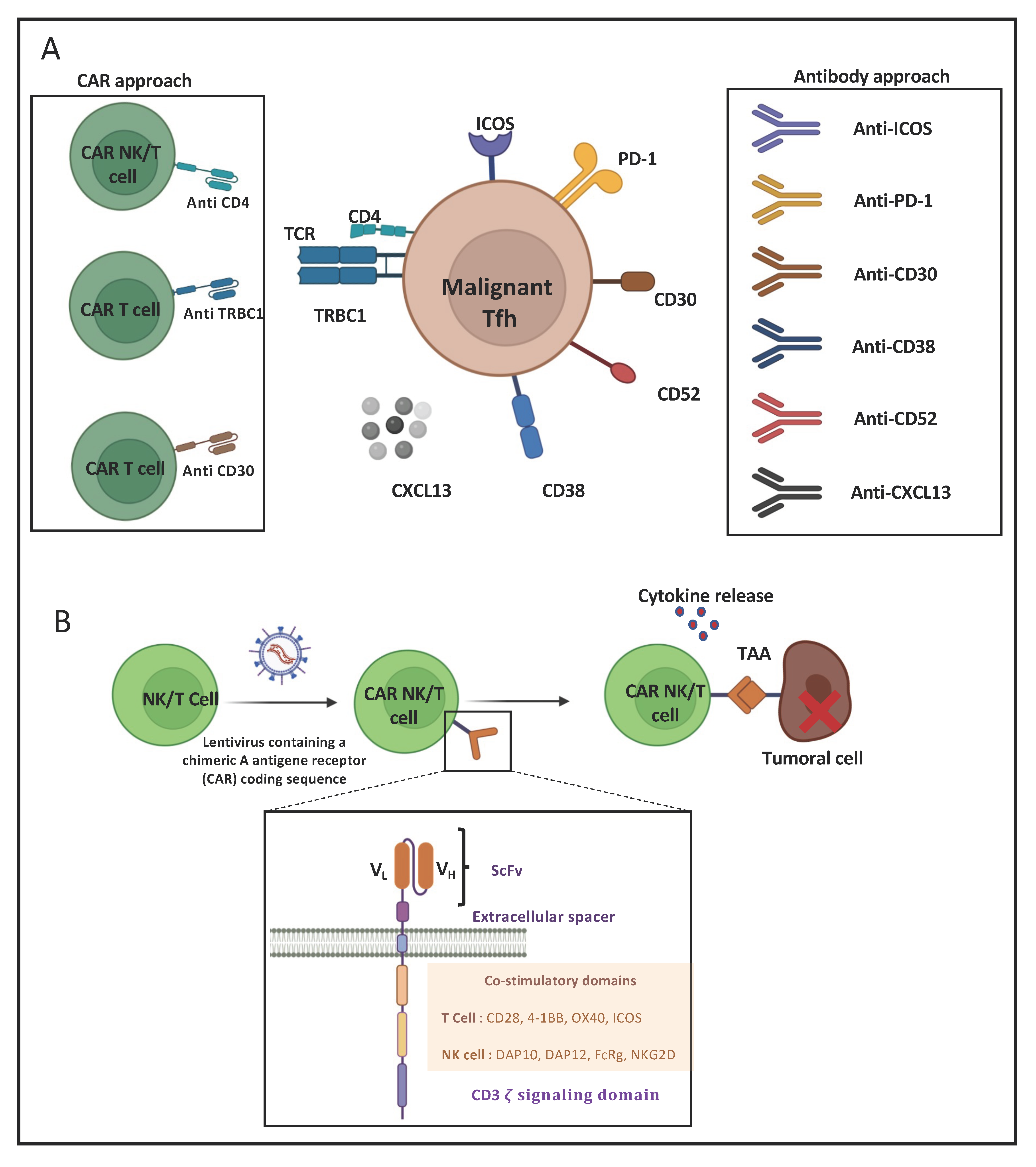
3.2. CAR-T-Cell-Based Immunotherapies for AITL Treatment
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390.
- de Leval, L.; Gisselbrecht, C.; Gaulard, P. Advances in the Understanding and Management of Angioimmunoblastic T-Cell Lymphoma. Br. J. Haematol. 2010, 148, 673–689.
- Laurent, C.; Baron, M.; Amara, N.; Haioun, C.; Dandoit, M.; Maynadié, M.; Parrens, M.; Vergier, B.; Copie-Bergman, C.; Fabiani, B.; et al. Impact of Expert Pathologic Review of Lymphoma Diagnosis: Study of Patients From the French Lymphopath Network. J. Clin. Oncol. 2017, 35, 2008–2017.
- Jaffe, E.; Arber, D.; Campo, E.; Harris, N. Quintanilla-Fend L. In Hematopathology e-Book; Elsevier: Amsterdam, The Netherlands, 2016.
- de Leval, L.; Parrens, M.; Le Bras, F.; Jais, J.-P.; Fataccioli, V.; Martin, A.; Lamant, L.; Delarue, R.; Berger, F.; Arbion, F.; et al. Angioimmunoblastic T-Cell Lymphoma Is the Most Common T-Cell Lymphoma in Two Distinct French Information Data Sets. Haematologica 2015, 100, e361–e364.
- Federico, M.; Rudiger, T.; Bellei, M.; Nathwani, B.N.; Luminari, S.; Coiffier, B.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Savage, K.J. Clinicopathologic Characteristics of Angioimmunoblastic T-Cell Lymphoma: Analysis of the International Peripheral T-Cell Lymphoma Project. J. Clin. Oncol. 2013, 31, 240.
- de Leval, L.; Rickman, D.S.; Thielen, C.; de Reynies, A.; Huang, Y.-L.; Delsol, G.; Lamant, L.; Leroy, K.; Brière, J.; Molina, T.; et al. The Gene Expression Profile of Nodal Peripheral T-Cell Lymphoma Demonstrates a Molecular Link between Angioimmunoblastic T-Cell Lymphoma (AITL) and Follicular Helper T (TFH) Cells. Blood 2007, 109, 4952–4963.
- Dobay, M.P.; Lemonnier, F.; Missiaglia, E.; Bastard, C.; Vallois, D.; Jais, J.-P.; Scourzic, L.; Dupuy, A.; Fataccioli, V.; Pujals, A.; et al. Integrative Clinicopathological and Molecular Analyses of Angioimmunoblastic T-Cell Lymphoma and Other Nodal Lymphomas of Follicular Helper T-Cell Origin. Haematologica 2017, 102, e148–e151.
- Palomero, T.; Couronné, L.; Khiabanian, H.; Kim, M.-Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent Mutations in Epigenetic Regulators, RHOA and FYN Kinase in Peripheral T Cell Lymphomas. Nat. Genet. 2014, 46, 166.
- Liang, P.-I.; Chang, S.-T.; Lin, M.-Y.; Hsieh, Y.-C.; Chu, P.-Y.; Chen, C.-J.; Lin, K.-J.; Jung, Y.-C.; Hwang, W.-S.; Huang, W.-T.; et al. Angioimmunoblastic T-Cell Lymphoma in Taiwan Shows a Frequent Gain of ITK Gene. Int. J. Clin. Exp. Pathol. 2014, 7, 6097–6107.
- Feldman, A.L.; Sun, D.X.; Law, M.E.; Novak, A.J.; Attygalle, A.D.; Thorland, E.C.; Fink, S.R.; Vrana, J.A.; Caron, B.L.; Morice, W.G.; et al. Overexpression of Syk Tyrosine Kinase in Peripheral T-Cell Lymphomas. Leukemia 2008, 22, 1139–1143.
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T Cell Activation. Annu. Rev. Immunol. 2009, 27, 591–619.
- Fujisawa, M.; Sakata-Yanagimoto, M.; Nishizawa, S.; Komori, D.; Gershon, P.; Kiryu, M.; Tanzima, S.; Fukumoto, K.; Enami, T.; Muratani, M.; et al. Activation of RHOA-VAV1 Signaling in Angioimmunoblastic T-Cell Lymphoma. Leukemia 2018, 32, 694–702.
- Vallois, D.; Dobay, M.P.D.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating Mutations in Genes Related to TCR Signaling in Angioimmunoblastic and Other Follicular Helper T-Cell–Derived Lymphomas. Blood 2016, 128, 1490–1502.
- Cortes, J.R.; Ambesi-Impiombato, A.; Couronné, L.; Quinn, S.A.; Kim, C.S.; da Silva Almeida, A.C.; West, Z.; Belver, L.; Martin, M.S.; Scourzic, L.; et al. RHOA G17V Induces T Follicular Helper Cell Specification and Promotes Lymphomagenesis. Cancer Cell 2018, 33, 259–273.e7.
- Vallois, D.; Dupuy, A.; Lemonnier, F.; Allen, G.; Missiaglia, E.; Fataccioli, V.; Ortonne, N.; Clavert, A.; Delarue, R.; Rousselet, M.-C.; et al. RNA Fusions Involving CD28 Are Rare in Peripheral T-Cell Lymphomas and Concentrate Mainly in Those Derived from Follicular Helper T Cells. Haematologica 2018, 103, e360–e363.
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA Mutation in Angioimmunoblastic T Cell Lymphoma. Nat. Genet. 2014, 46, 171–175.
- Rohr, J.; Guo, S.; Huo, J.; Bouska, A.; Lachel, C.; Li, Y.; Simone, P.D.; Zhang, W.; Gong, Q.; Wang, C.; et al. Recurrent Activating Mutations of CD28 in Peripheral T-Cell Lymphomas. Leukemia 2016, 30, 1062–1070.
- Ohmoto, A.; Fuji, S. Cyclosporine for Angioimmunoblastic T-Cell Lymphoma: A Literature Review. Expert Rev. Hematol. 2019, 12, 975–981.
- Boddicker, R.L.; Razidlo, G.L.; Dasari, S.; Zeng, Y.; Hu, G.; Knudson, R.A.; Greipp, P.T.; Davila, J.I.; Johnson, S.H.; Porcher, J.C.; et al. Integrated Mate-Pair and RNA Sequencing Identifies Novel, Targetable Gene Fusions in Peripheral T-Cell Lymphoma. Blood 2016, 128, 1234–1245.
- Liu, Y.; Wang, X.; Deng, L.; Ping, L.; Shi, Y.; Zheng, W.; Lin, N.; Wang, X.; Tu, M.; Xie, Y.; et al. ITK Inhibition Induced in Vitro and in Vivo Anti-Tumor Activity through Downregulating TCR Signaling Pathway in Malignant T Cell Lymphoma. Cancer Cell Int. 2019, 19, 32.
- Streubel, B.; Vinatzer, U.; Willheim, M.; Raderer, M.; Chott, A. Novel t(5;9)(Q33;Q22) Fuses ITK to SYK in Unspecified Peripheral T-Cell Lymphoma. Leukemia 2006, 20, 313–318.
- Pechloff, K.; Holch, J.; Ferch, U.; Schweneker, M.; Brunner, K.; Kremer, M.; Sparwasser, T.; Quintanilla-Martinez, L.; Zimber-Strobl, U.; Streubel, B.; et al. The Fusion Kinase ITK-SYK Mimics a T Cell Receptor Signal and Drives Oncogenesis in Conditional Mouse Models of Peripheral T Cell Lymphoma. J. Exp. Med. 2010, 207, 1031–1044.
- Dierks, C.; Adrian, F.; Fisch, P.; Ma, H.; Maurer, H.; Herchenbach, D.; Forster, C.U.; Sprissler, C.; Liu, G.; Rottmann, S.; et al. The ITK-SYK Fusion Oncogene Induces a T-Cell Lymphoproliferative Disease in Mice Mimicking Human Disease. Cancer Res. 2010, 70, 6193–6204.
- Lechner, K.S.; Neurath, M.F.; Weigmann, B. Role of the IL-2 Inducible Tyrosine Kinase ITK and Its Inhibitors in Disease Pathogenesis. J. Mol. Med. 2020, 98, 1385–1395.
- Zhong, Y.; Dong, S.; Strattan, E.; Ren, L.; Butchar, J.P.; Thornton, K.; Mishra, A.; Porcu, P.; Bradshaw, J.M.; Bisconte, A.; et al. Targeting Interleukin-2-Inducible T-Cell Kinase (ITK) and Resting Lymphocyte Kinase (RLK) Using a Novel Covalent Inhibitor PRN694. J. Biol. Chem. 2015, 290, 5960–5978.
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.-M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib Is an Irreversible Molecular Inhibitor of ITK Driving a Th1-Selective Pressure in T Lymphocytes. Blood 2013, 122, 2539–2549.
- Kumar, A.; Vardhana, S.; Moskowitz, A.J.; Porcu, P.; Dogan, A.; Dubovsky, J.A.; Matasar, M.J.; Zhang, Z.; Younes, A.; Horwitz, S.M. Pilot Trial of Ibrutinib in Patients with Relapsed or Refractory T-Cell Lymphoma. Blood Adv. 2018, 2, 871–876.
- Yoo, H.Y.; Kim, P.; Kim, W.S.; Lee, S.H.; Kim, S.; Kang, S.Y.; Jang, H.Y.; Lee, J.-E.; Kim, J.; Kim, S.J.; et al. Frequent CTLA4-CD28 Gene Fusion in Diverse Types of T-Cell Lymphoma. Haematologica 2016, 101, 757–763.
- Lee, G.J.; Jun, Y.; Jeon, Y.K.; Lee, D.; Lee, S.; Kim, J. Mice Transgenic for Human CTLA4-CD28 Fusion Gene Show Proliferation and Transformation of ATLL-like and AITL-like T Cells. Oncoimmunology 2022, 11, 2015170.
- Nguyen, T.B.; Sakata-Yanagimoto, M.; Fujisawa, M.; Nuhat, S.T.; Miyoshi, H.; Nannya, Y.; Hashimoto, K.; Fukumoto, K.; Bernard, O.A.; Kiyoki, Y. Dasatinib Is an Effective Treatment for Angioimmunoblastic T-Cell Lymphoma. Cancer Res. 2020, 80, 1875–1884.
- Mondragón, L.; Mhaidly, R.; De Donatis, G.M.; Tosolini, M.; Dao, P.; Martin, A.R.; Pons, C.; Chiche, J.; Jacquin, M.; Imbert, V.; et al. GAPDH Overexpression in the T Cell Lineage Promotes Angioimmunoblastic T Cell Lymphoma through an NF-ΚB-Dependent Mechanism. Cancer Cell 2019, 36, 268–287.e10.
- Kridin, K.; Ahmed, A.R. Post-Rituximab Immunoglobulin M (IgM) Hypogammaglobulinemia. Autoimmun. Rev. 2020, 19, 102466.
- Stone, E.L.; Pepper, M.; Katayama, C.D.; Kerdiles, Y.M.; Lai, C.-Y.; Emslie, E.; Lin, Y.C.; Yang, E.; Goldrath, A.W.; Li, M.O.; et al. ICOS Coreceptor Signaling Inactivates the Transcription Factor FOXO1 to Promote Tfh Cell Differentiation. Immunity 2015, 42, 239–251.
- Weber, J.P.; Fuhrmann, F.; Feist, R.K.; Lahmann, A.; Al Baz, M.S.; Gentz, L.-J.; Vu Van, D.; Mages, H.W.; Haftmann, C.; Riedel, R.; et al. ICOS Maintains the T Follicular Helper Cell Phenotype by Down-Regulating Krüppel-like Factor 2. J. Exp. Med. 2015, 212, 217–233.
- Warnatz, K.; Bossaller, L.; Salzer, U.; Skrabl-Baumgartner, A.; Schwinger, W.; van der Burg, M.; van Dongen, J.J.M.; Orlowska-Volk, M.; Knoth, R.; Durandy, A.; et al. Human ICOS Deficiency Abrogates the Germinal Center Reaction and Provides a Monogenic Model for Common Variable Immunodeficiency. Blood 2006, 107, 3045–3052.
- Shi, J.; Hou, S.; Fang, Q.; Liu, X.; Liu, X.; Qi, H. PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity 2018, 49, 264–274.
- Good-Jacobson, K.L.; Szumilas, C.G.; Chen, L.; Sharpe, A.H.; Tomayko, M.M.; Shlomchik, M.J. PD-1 Regulates Germinal Center B Cell Survival and the Formation and Affinity of Long-Lived Plasma Cells. Nat. Immunol. 2010, 11, 535–542.
- Han, L.; Liu, F.; Li, R.; Li, Z.; Chen, X.; Zhou, Z.; Zhang, X.; Hu, T.; Zhang, Y.; Young, K.; et al. Role of Programmed Death Ligands in Effective T-Cell Interactions in Extranodal Natural Killer/T-Cell Lymphoma. Oncol. Lett. 2014, 8, 1461–1469.
- Fiore, D.; Cappelli, L.V.; Broccoli, A.; Zinzani, P.L.; Chan, W.C.; Inghirami, G. Peripheral T Cell Lymphomas: From the Bench to the Clinic. Nat. Rev. Cancer 2020, 20, 323–342.
- Wartewig, T.; Kurgyis, Z.; Keppler, S.; Pechloff, K.; Hameister, E.; Öllinger, R.; Maresch, R.; Buch, T.; Steiger, K.; Winter, C.; et al. PD-1 Is a Haploinsufficient Suppressor of T Cell Lymphomagenesis. Nature 2017, 552, 121–125.
- Rauch, D.A.; Conlon, K.C.; Janakiram, M.; Brammer, J.E.; Harding, J.C.; Ye, B.H.; Zang, X.; Ren, X.; Olson, S.; Cheng, X.; et al. Rapid Progression of Adult T-Cell Leukemia/Lymphoma as Tumor-Infiltrating Tregs after PD-1 Blockade. Blood 2019, 134, 1406–1414.
- Barta, S.K.; Zain, J.; MacFarlane, A.W.; Smith, S.M.; Ruan, J.; Fung, H.C.; Tan, C.R.; Yang, Y.; Alpaugh, R.K.; Dulaimi, E.; et al. Phase II Study of the PD-1 Inhibitor Pembrolizumab for the Treatment of Relapsed or Refractory Mature T-Cell Lymphoma. Clin. Lymphoma Myeloma Leuk 2019, 19, 356–364.e3.
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704.
- Neuwelt, A.; Al-Juhaishi, T.; Davila, E.; Haverkos, B. Enhancing Antitumor Immunity through Checkpoint Blockade as a Therapeutic Strategy in T-Cell Lymphomas. Blood Adv. 2020, 4, 4256–4266.
- Zhang, L.; Mai, W.; Jiang, W.; Geng, Q. Sintilimab: A Promising Anti-Tumor PD-1 Antibody. Front. Oncol. 2020, 10, 594558.
- Guo, Y.M.; Liu, X.F.; Jiao, L.J.; Yin, S.Y.; Wang, Z.; Li, X.X.; Ma, Z.P.; Yang, J.M.; He, M.X. Angioimmunoblastic T-cell lymphoma: Histopathological grading and prognosis. Zhonghua Bing Li Xue Za Zhi 2019, 48, 784–790.
- Feng, X.; Guo, W.; Wang, Y.; Li, J.; Zhao, Y.; Qu, L.; Yan, X.; Li, J.; Guo, Q.; Young, K.H.; et al. The Short-Term Efficacy and Safety of Brentuximab Vedotin Plus Cyclophosphamide, Epirubicin and Prednisone in Untreated PTCL: A Real-World, Retrospective Study. Adv. Ther. 2022, 39, 532–543.
- Sabattini, E.; Pizzi, M.; Tabanelli, V.; Baldin, P.; Sacchetti, C.S.; Agostinelli, C.; Zinzani, P.L.; Pileri, S.A. CD30 Expression in Peripheral T-Cell Lymphomas. Haematologica 2013, 98, e81–e82.
- Salles, G.; Barrett, M.; Foà, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273.
- Brodfuehrer, J.; Rankin, A.; Edmonds, J.; Keegan, S.; Andreyeva, T.; Lawrence-Henderson, R.; Ozer, J.; Gao, H.; Bloom, L.; Boisvert, A.; et al. Quantitative Analysis of Target Coverage and Germinal Center Response by a CXCL13 Neutralizing Antibody in a T-Dependent Mouse Immunization Model. Pharm. Res. 2014, 31, 635–648.
- Bhamidipati, K.; Silberstein, J.L.; Chaichian, Y.; Baker, M.C.; Lanz, T.V.; Zia, A.; Rasheed, Y.S.; Cochran, J.R.; Robinson, W.H. CD52 Is Elevated on B Cells of SLE Patients and Regulates B Cell Function. Front. Immunol. 2020, 11, 626820.
- Jiang, L.; Yuan, C.M.; Hubacheck, J.; Janik, J.E.; Wilson, W.; Morris, J.C.; Jasper, G.A.; Stetler-Stevenson, M. Variable CD52 Expression in Mature T Cell and NK Cell Malignancies: Implications for Alemtuzumab Therapy. Br. J. Haematol. 2009, 145, 173–179.
- Wulf, G.G.; Altmann, B.; Ziepert, M.; D’amore, F.; Held, G.; Greil, R.; Tournilhac, O.; Relander, T.; Viardot, A.; Wilhelm, M. Alemtuzumab plus CHOP versus CHOP in Elderly Patients with Peripheral T-Cell Lymphoma: The DSHNHL2006-1B/ACT-2 Trial. Leukemia 2021, 35, 143–155.
- Buckstein, R.; Fraser, G.; Cheung, M.; Kukreti, V.; Kuruvilla, J.; Imrie, K.; Piliotis, E.; Pond, G.; Windsor, J.; Ghorab, Z.; et al. Alemtuzumab and CHOP Chemotherapy for the Treatment of Aggressive Histology Peripheral T Cell Lymphomas: A Multi-Center Phase I Study. Clin. Lymphoma Myeloma Leuk 2016, 16, 18–28.e4.
- Krug, A.; Martinez-Turtos, A.; Verhoeyen, E. Importance of T, NK, CAR T and CAR NK Cell Metabolic Fitness for Effective Anti-Cancer Therapy: A Continuous Learning Process Allowing the Optimization of T, NK and CAR-Based Anti-Cancer Therapies. Cancers 2021, 14, 183.
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459.
- Mhaidly, R.; Krug, A.; Gaulard, P.; Lemonnier, F.; Ricci, J.-E.; Verhoeyen, E. New Preclinical Models for Angioimmunoblastic T-Cell Lymphoma: Filling the GAP. Oncogenesis 2020, 9, 73.
- Breman, E.; Demoulin, B.; Agaugué, S.; Mauën, S.; Michaux, A.; Springuel, L.; Houssa, J.; Huberty, F.; Jacques-Hespel, C.; Marchand, C.; et al. Overcoming Target Driven Fratricide for T Cell Therapy. Front. Immunol. 2018, 9, 2940.
- Scarfò, I.; Ormhøj, M.; Frigault, M.J.; Castano, A.P.; Lorrey, S.; Bouffard, A.A.; van Scoyk, A.; Rodig, S.J.; Shay, A.J.; Aster, J.C.; et al. Anti-CD37 Chimeric Antigen Receptor T Cells Are Active against B- and T-Cell Lymphomas. Blood 2018, 132, 1495–1506.
- Pinz, K.G.; Yakaboski, E.; Jares, A.; Liu, H.; Firor, A.E.; Chen, K.H.; Wada, M.; Salman, H.; Tse, W.; Hagag, N.; et al. Targeting T-Cell Malignancies Using Anti-CD4 CAR NK-92 Cells. Oncotarget 2017, 8, 112783–112796.
- Maciocia, P.M.; Wawrzyniecka, P.A.; Philip, B.; Ricciardelli, I.; Akarca, A.U.; Onuoha, S.C.; Legut, M.; Cole, D.K.; Sewell, A.K.; Gritti, G.; et al. Targeting the T Cell Receptor β-Chain Constant Region for Immunotherapy of T Cell Malignancies. Nat. Med. 2017, 23, 1416–1423.


