1. Introduction
Peripheral T-cell lymphomas (PTCL) are a diverse group of overall rare and aggressive lymphomas that develop from the oncogenic transformation of mature (i.e., post-thymic) T cells. PTCLs account for less than 10% of non-Hodgkin lymphomas worldwide. The occurrence of the different subtypes is affected by several factors, such as age, gender, ethnic origin, genetics and immunological disorders. The last WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues recognized 30 distinct PTCL subtypes, which are grouped according to their main clinical presentations
[1].
Among these various PTCL subtypes, T follicular helper (Tfh) PTCLs have been recently recognized as the most frequent PTCL entity in European countries, encompassing more than 30% of non-cutaneous PTCL
[2][3][2,3].
Angioimmunoblastic T-cell lymphoma (AITL) is the most frequent and was the first Tfh PTCL identified. Of importance, PTCL and, thus, AITL remain rare cancers compared to B-cell lymphomas. This is an aggressive disease with poor clinical outcomes, the 5-year overall survival being around 30% and preferentially affecting patients over the age of 60. It is a systemic disease frequently associated with generalized lymphadenopathy, hepatosplenomegaly, cutaneous rash and serous involvement. Hypergammaglobulinemia and autoimmune disorders, such as autoimmune cytopenia, are frequent manifestations and could reflect the Tfh derivation of this lymphoma
[4]. At the pathological level, AITL is characterized by a polymorphic infiltrate that usually destroys the lymph node architecture, composed of neoplastic T cells with a Tfh phenotype and a prominent tumor microenvironment (TME). This microenvironment is a mix of reactive hematopoietic cells, including T cells, plasma cells, eosinophils, B cells and immunoblasts, which are large B cells that are frequently EBV-positive; and stromal cells, including post-capillary venule hyperplasia and follicular dendritic cell expansion
[5]. Neoplastic T cells have irregular nuclei and clear cytoplasm. They are CD4+ T cells, and they express the Tfh-associated molecules CD10, C-X-C motif chemokine ligand 13 (CXCL13), B-cell lymphoma 6 (BCL6), programmed death-1 (PD-1), C-X-C motif chemokine receptor 5 (CXCR5) and inducible co-stimulatory (ICOS) molecules
[6]. Gene expression studies showed an enriched Tfh signature in AITL that resembled their normal Tfh counterparts
[7].
In addition to AITL, follicular PTCL, a rare PTCL subtype where neoplastic cells have a follicular pattern and a part of PTCL not otherwise specified (PTCL-NOS), representing around 25% of PTCL NOS, expresses Tfh markers. AITL, follicular PTCL and nodal PTCL with a Tfh phenotype present similar clinical presentations, gene expression profiles, DNA copy numbers, anomalies and mutational profiles, suggesting they are closely related
[8]. These three subsets were gathered under the same umbrella entity Tfh PTCL in the latest WHO classification
[1].
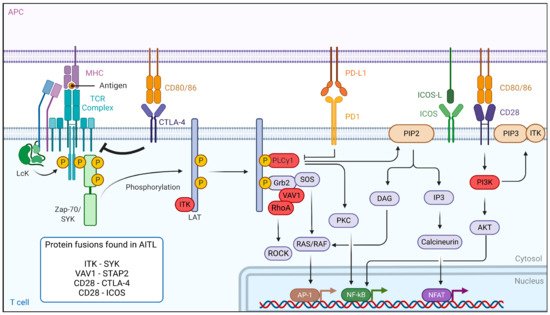
2. Dysregulation of the TCR Signaling Pathway in AITL Reveals New Treatment Options
In PTCL and AITL patients, mutations in components of the TCR signaling pathway, causing constitutive activation of the TCR, probably play a major role in T-cell pathogenesis
[9][10][11][20,54,55]. The binding of the TCR to antigen-presenting cells leads to a cascade of events. First, the activation of the Src kinase LCK leads to the binding of zeta-chain-associated protein kinase 70 (ZAP-70) and IL-2-inducible T-cell kinase (ITK), which are then phosphorylated and activated (
Figure 13). ZAP-70 then phosphorylates its targets, the adaptors linker for activation of T cells (LAT), SH2-domain-containing leukocyte protein (SLP-76) and phospholipase Cγ1 (PLCγ1), which serve as a platform for the recruitment of ITK, vav guanine nucleotide exchange factor 1 (Vav1) and the non-catalytic region of tyrosine kinase adaptor protein 1 (Nck1) in order to build a T-cell signaling complex. After its phosphorylation, PLCγ1 hydrolyzes phosphatidylinositol-4,5-biphosphate (PIP2) to produce dacylglycerol and inositol 3 phosphate (IP3), leading to the activation of nuclear factor of activated T cells (NFAT). In parallel, co-stimulation through CD28 leads to PI3K activation and PIP3 accumulation in the cells. PIP3 binds to ITK and forms a signaling complex at the cell membrane. The activation of the TCR also modulates other downstream signaling pathways, including the PI3K, NF-κB, MAPK and GTPase-dependent pathways
[12][56].
Figure 13. TCR-signaling and co-stimulatory pathways affected in AITL. Peptides/antigens bind to the MHC-class molecules and engage the TCR. The TCR signaling strength depends on the co-stimulatory molecules, e.g., CD28 or CTLA-4. This is followed by a series of events leading to the phosphorylation of the different components of the TCR complex. Genes mutated in the TCR-signaling pathway in AITL are indicated in red and mostly lead to antigen-independent hyper-activation of TCR signaling. Aberrant fusion proteins of the TCR signaling pathway are often encountered in AITL and are indicated in the box. Figure generated by
Biorender.com.
The RHOA
G17V mutant is found frequently (70%) in AITL malignant cells. This mutation drives Tfh differentiation, but it can also bind Vav1, resulting in Vav1 hyperphosphorylation
[13][57] and NFAT signaling activation, inducing T-cell proliferation and transformation
[14][15][16][10,18,23]. This indicates that the RHOA
G17V mutation could be a major player in T-cell signaling activation. For this reason, multikinase inhibitors (desatinib) or PI3K inhibitors (duvelisib) are proposed as therapeutic options
[9][17][19,20]. Additionally, many other components of the TCR signaling pathways, such as phospholipase C
γ1 (14%)
[14][10], CD28 (9–11%)
[16][18][23,58], Src family tyrosine kinase (FYN) (3–4%)
[14][10] and Vav1 itself (50%) are mutated in AITL. Moreover, a VAV1-STAP2 fusion protein is detected in some cases. To therapeutically interfere with the antigen-independent strong TCR signaling caused by these mutations, one option is to use calcineurin inhibitors, such as cyclosporine 1
[19][59]. Authors reviewed several clinical trials and extracted a total of 26 patients with AITL receiving cyclosporine. Interestingly, the overall response rate in AITL patients was impressive (86%), suggesting the utility of using calcineurin inhibitors in already-treated AITL cases
[19][59]. However, the authors do insist that prudence is warranted in the interpretation and comparison of different clinical studies, especially because of the small number of AITL cases. Finally, in the case of mutated Vav1, Rac 1 inhibitors could also be an option
[20][60].
Importantly, ITK is highly expressed in AITL patients. One study reports that ITK was highly phosphorylated in 70% of the AITL patients, and this correlated with a poor response to first-line treatment
[21][61]. Additionally, spleen-associated tyrosine kinase (SYK), normally expressed in B cells, demonstrated an aberrantly high expression in the majority of AITL patients
[22]. Therefore, SYK inhibitors might be promising therapeutic agents. In addition, translocation in some PTCL patients, including AITL, resulted in the ITK-SYK and ITK-FER fusions
[22][23][22,62], which induced, independent of antigen binding, phosphorylation of TCR proximal proteins and acted as strong oncogenic drivers in T-cell lymphoma in mouse models
[22][23][24][22,62,63]. Therefore, ITK is a target for the design of new candidates for targeted therapies, especially since it was shown that ITK inhibition leads to a decrease in the invasion and migration of malignant T cells
[21][25][61,64]. This suggests that ITK inhibitor treatment might be effective in ITK-positive AITL patients since preclinical data showed anti-cancer activity in B- and T-cell leukemia
[26][65]. Evidently, SYK inhibitors could also be proposed in the case of ITK-SYK fusion
[22]. A combination of an ITK inhibitor with CHOP may be a promising therapeutic regimen. Ibrutinib was also evaluated for T-cell lymphoma since it was shown to be specific not only for BTK (Bruton tyrosine kinase) but also inhibited ITK
[27][28][66,67]. Ibrutinib was tested in a clinical trial enrolling T-cell lymphoma patients. However, this only resulted in an overall response rate of 8% in these patients. Currently, CPI-818, a new selective ITK inhibitor is being tested in a phase I clinical trial including patients with refractory T-cell lymphoma, but no results are currently available (NCT03952078).
As mentioned above, an efficient immune response relies on TCR costimulatory molcules providing a secondary signal upon TCR engagement with an antigen. CD28 is one of these co-stimulation receptors expressed at the T-cell surface and is mutated in 10% of AITL patients. Two specific CD28 mutations (D124 and T195) result in extended activation of the T-cell receptor because they have a stronger affinity for their ligand than the WT CD28 and induce signaling pathways implicated in cytokine production and T-cell proliferation
[18][58]. Moreover, the CD28-ICOS and CD28-CTLA4 gene fusions were detected in AITL
[14][29][10,68]. Unexpectedly, the CD28-CTLA4 fusion gene can convert the normal inhibitory signals induced by CTLA4 stimulation in hyperactive CD28 signaling in AITL T cells
[29][68]. Recently, a mouse model expressing the CTLA4-CD28 fusion exclusively in a T-cell lineage was generated, and these mice developed an AITL-like lymphoma, underlining the CTLA4-CD28 transforming capacity
[30][69]. Nguyen et al.
[31][70], using a genetic mouse model for AITL (TET2
−/−; RHOA
G17V), demonstrated that dasatinib, a multikinase inhibitor, inhibited hyperactivated TCR signaling in these mice and increased their survival. Additionally, they report a clinical phase I trial including five AITL patients that confirmed a high response rate to dasatinib (UMIN000025856). These results need to be taken with care since the small number of patients limited the relevance of the study. Another phase I/II trial is ongoing using dasatinib. Importantly, the same drug also blocked the RhoA-Vav1 TCR signaling in AITL
[13][57]. Alternatively, in the case of CTLA4-CD28, an anti-CTLA4 immunotherapy (ipilimumab) is proposed
[29][68].
FYN is a tyrosine kinase that also plays an essential role in T-cell activation. Multiple mutations in FYN, which invalidate the inhibitory function of the FYN domain SH2 and result in a constitutive activation of the tyrosine kinase and T-cell activation
[9][20] were identified in AITL patients. Many more mutations in PLCG1, CARD11 and CTNNB1 as well as proteins involved in the PI3K or MAPK pathways, which are implicated in the stabilization of stimulatory signals in T cells, have been described in AITL. Interestingly, CARD11 mutations seem to be at the origin of constitutive NF-
κB signaling and might assist in tumor outgrowth
[14][10]. It was actually the NF-
κB pathway that was shown to be constitutively upregulated in one of the recent preclinical AITL mouse models (
[32][41]).
3. Immunotherapeutic Approaches for AITL
Escaping the immune system is the main characteristic of a tumor cell. In order to limit the anarchic proliferation of cancer cells, chemotherapies are often used in tumors that have no specific treatment yet, such as AITL. These therapies are proposed to limit the proliferation of dividing cells, i.e., healthy cells as well as tumor cells. This explains the poor benefits of such treatment and the major risk of relapse or even the absence of signs of remission. Importantly, targeting a specific cancer cell type without inducing an effect on healthy cells requires an in-depth knowledge of its phenotype and its surface markers, which can serve as an asset for the development of specific therapies, in particular immunotherapies. If some healthy cells are affected, it is called on-target off-target effects because certain cancer epitopes are partially expressed on non-malignant cells. These therapies include, for example, monoclonal antibodies and chimeric antigen receptors.
3.1. Monoclonal-Antibody-Based Immunotherapies for AITL Treatment
Monoclonal antibodies aim either to deplete the target by inducing antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity or the direct triggering of apoptosis leading to cell death
[33][71]. However, they can also function by blocking a cell–cell interaction or a pathway favorable to tumor cell development, proliferation or survival. The CD4+ Tfh cell, the malignant driver of AITL, highly expresses surface markers, such as CXCR5, PD-1, ICOS and CD40L, that determine its phenotype
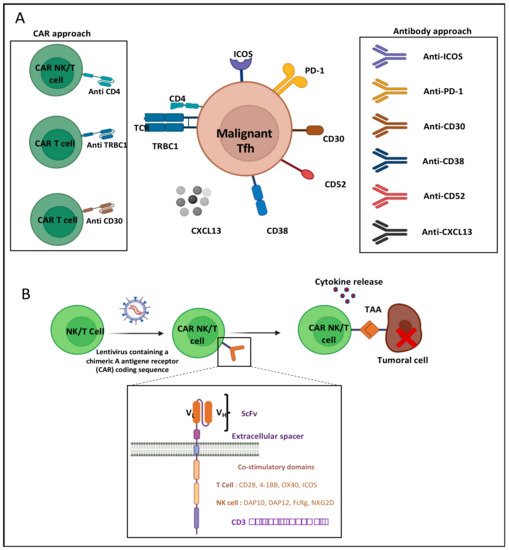
[7] and can serve as targets for immunotherapy (
Figure 24A). Indeed, the CD4+ Tfh cells play an important role in B-cell maturation within the germinal center towards GC B cells. Their survival is highly dependent on ICOS surface expression, which is induced by TCR stimulation
[34][72]. Moreover, ICOS plays a crucial role in Tfh localization
[35][73]. Once Tfh cells leave the T zone and migrate to the germinal center, ICOS boosts the cognate T- to B-cell interaction and thereby enables an optimal antibody-based immune response
[36][74]. AITL Tfh CD4 cells show very high similarity to healthy Tfh cells in their T-cell–B-cell interaction through ICOS-ICOSL binding. Thus, ICOS was considered a possible target for immunotherapy. Currently, a phase I clinical trial targeting ICOS by the MEDI-570 antibody is ongoing, in which recurrent AITL patients were included among other peripheral T-cell lymphomas (NCT02520791). The expected outcome of this trial is a block in Tfh growth and tumor progression, but no clear beneficial results are available, though the treatment does not seem to be toxic.
Figure 24. AITL immunotherapy approaches. (A) T-cell follicular helper (TFH) cells of AITL express several surface markers targetable by antibodies and CARs: inducible T-cell co-stimulator (ICOS), programed death 1 (PD-1), cluster of differentiation (CD) CD4, CD30, CD38, CD52 and TRBC1. Neutralizing antibodies against chemokine 13 (CXCL13) can also be used to inhibit TFH migration to germinal center. (B) Design of CAR-NK/T-cell generation and tumor targeting. Natural killer (NK) or T cells are transduced with a lentivirus to express a chimeric antigen receptor (CAR). NK/T cells will, via the single chain variable fragment (scFv) exposed by the CAR, recognize the tumor-associated antigen (TAA), allowing NK/T-cell activation through the CAR signaling and co-stimulation domains. VL: variable light chain. VH: variable heavy chain.
As ICOS, PD-1 is a B7 family molecule highly expressed by Tfh. While PD-1 is known to be inhibitory in immune cells, PD-1 plays an important role in Tfh development and activity. The PD-1 interaction with its ligand PDL1, which is expressed by B cells, favors the follicular recruitment of T cells expressing ICOS at the T–B border
[37][75]. Within the germinal center, PD-1 expression controls Tfh and B-cell proliferation and survival
[38][76]. In addition, PD-1 was found to inhibit the cytotoxic function by interacting with the PDLs expressed on anti-tumoral cells, such as natural killers and cytotoxic CD8 cells in lymphoma
[39][77]. Taken together, PD-1 presented an important candidate target for immunotherapy in AITL. In this context, Mondragon et al. used an anti-PD-1 antibody in combination with a non-canonical NF-kB inhibitor to treat mice bearing AITL tumors. Survival increased up to 70% compared to non-treated mice
[32][41]. These are encouraging results, but the same
res
earchtudy showed only 40% survival upon anti-PD-1 as a single treatment. Fiore et al.
[40][78] mentioned that pembrolizumab and nivolumab (humanized anti-PD-1 antibodies) were effective in different lymphoma subtypes. Pembrolyzumab was highly effective in relapsed or refractory NK/T-cell lymphoma patients, but blockade of the PD-1 axis is still disputable in other lymphomas subtypes
[40][78]. It was also believed that in PTCL PD-1 itself could be a tumor suppressor
[41][79] suggesting that anti-PD-1 could lead to exactly the opposite of what is intended, meaning increased lymphoma. Indeed, in some mouse studies, anti-PD-1 treatment caused a violent progression of adult T-cell lymphomas
[42][80]. At present, trials including multiple types of PTCL have shown only very low activity upon anti-PD-1 as a single treatment
[43][44][81,82]. A combination of checkpoint inhibitors with other agents might enhance the anti-tumor activity in T-cell lymphoma
[45][83]. Importantly, clinical studies showed that classic HDAC inhibitors improved AITL patient ORR over 33–50% but enhanced PD-1 expression. An ongoing phase II clinical trial is testing Chilamide (HDAC inhibitor) combined with Sinlimab
[46][84], a stable anti-PD-1 antibody (NCT04831710).
The T-cell follicular helper (Tfh) origin of AITL expresses several surface markers: inducible T-cell co-stimulator (ICOS), programed death 1 (PD-1), cluster of differentiation (CD) CD4, CD30, CD38, CD52 and TRBC1. Antibodies and CAR-NK/T cells target these extracellular markers. Indeed, a surface target was revealed in a cohort of 62 AITL patients, which showed that CD38-positive AITL cell presence was a risk factor associated with reduced overall survival
[47][85]. An anti-CD38 antibody, Daratumuab is already approved for myeloma treatment. Moreover, a phase II clinical trial is ongoing to treat AITL patients with Daratumuab. However, it is not administered as a single agent but in combination with chemotherapies (NCT04251065). In AITL, CD30 surface expression can be detected in up to 43% of the patients
[48][86]. CD30 is a transmembrane glycoprotein that has a pleiotropic effect on cell growth and survival
[49][87]. Brentuximab Vedotin is an anti-CD30 antibody approved by the FDA in relapsed and refractory CD30+ PTCL, as recently the ECHELON-2 trial showed a statistical improvement in the response rate for 83% of patients. However, an AITL subgroup analysis did not indicate an improvement for these patients. Currently, a phase II clinical trial is recruiting to test Brentuximab Vedotin as a therapy for AITL patients among other lymphoma subtypes (NCT02588651). Since in AITL the vast majority of the B-cell component of the lymphoma consists of GC B cells expressing the CD20 mature B-cell marker, CD20 was considered to be an indirect target for immunotherapy in AITL. The T- and B-cell interaction in AITL is indeed important for their mutual survival. Retuximab is an antibody targeting CD20 that provided high efficacy in B-cell lymphoma
[50][88]. Currently, a clinical trial is testing Retuximab in relapsed/refractory AITL patients combined with lenalidomide, a potent anti-angiogenesis and a neoplasm-bocking antibody (NCT04319601). Moreover, CXCR5 is highly expressed by Tfh cells in AITL and binds the chemokine CXCL13, which allows follicular homing and is a chemoattractant for B cells. The neutralizing antibody anti-chemokine 13 (CXCL13) may lead to a reduction in B-cell recruitment in the germinal center as suggested by Brodfuehrer et al. in a mouse model
[51][89]. However, no proof of its efficacy to treat AITL malignancy has been reported.
Finally, CD52 is considered as an immunotherapy target since it is a glycophosphatidylinositol that is widely expressed by the immune system and, thus, by both B and T cells. This cell surface protein plays an important role in T-cell homeostasis, immunosuppression and NF-
κB inhibition
[52][90]. CD52 is used as target for T- and NK-cell malignancies, including AITL, in which CD52 is highly expressed
[53][91]. Therefore, several clinical trials have been initiated using Alemtuzumab (CD52 antibody)
[54][55][28,92]. Currently a clinical trial is using Alemtuzumab in addition to dose dense CHOP (A-CHOP-14) to treat PTCL, including AITL patients (NCT00725231). However, in several trials this drug treatment revealed high toxicity and does not show a clear beneficial effect.
3.2. CAR-T-Cell-Based Immunotherapies for AITL Treatment
CAR-cell therapy consists of genetically modifying a natural killer (NK) or a T cell so they can express a chimeric antigen receptor (CAR). These synthetic receptors allow the CAR-NK or -T cells to recognize a tumor-associated antigen and eliminate the cancer cells that escaped the immune system (
Figure 24A,B). The CAR structure is similar to a T-cell receptor (TCR). Nowadays, five CAR generations exist, depending on their intracellular functional domains
[56][93]. The most commonly used CAR-T cells are already accepted by the FDA for patient treatment and contain the CD28 and/or the 4-1BB co-stimulatory domains and the CD3ζ signaling chain. The choice of a co-stimulatory signal influences the T-cell metabolic status and persistence in vivo
[56][93]. The extracellular part is composed of a single-chain variable fragment (scFv) made of a light and heavy variable region of an antibody (VL and VH) that senses and binds to a target antigen. CAR-T-cell treatment gave spectacular results in B-cell malignancies treated with anti-CD19 CAR-T cells
[57][94]. Several clinical trials are ongoing to extend this strategy to different lymphoma subtypes. Many ongoing CAR-NK/T-cell therapies are being evaluated in clinical trials recruiting PTCL patients, including AITL, because of common surface targets. The recent generation of AITL mouse models very closely recapitulating human disease
(see Section 3 and Section 5) could allow advances in the testing of CAR-T/NK-cell approaches
[58][17]. However, it needs to be mentioned that T-cell lymphomas do not express surface markers that are excluded from healthy T cells. Therefore, the choice of the CAR-specific target is not an easy one because, as a side-effect, the CAR-T cells could attack themselves through T-cell fratricide, a mechanism originally revealed to maintain T-cell homeostasis. CAR-T cells expressing the fusion with CD3ζ acquire a specificity for ligands expressed on hematological and solid cancers. However, these ligands or receptors can also be transiently or permanently expressed by activated T cells, implying that CAR-T cells may undergo self-killing (fratricide) during CAR-T-cell production
[59][95]. This might hinder therapeutic efficiency since T-cell fratricide might prevent the production of the required quantities of T cells for clinical applications. This is particularly true when the CAR target itself is specific for the T-cell lineage (CD4, CD7 or CD5) in order to eliminate T-cell leukemic cells. Therefore, research is conducted to avoid this unwanted effect
[59][60][95,96].
AITL Tfh cells are derived from CD4 T cells. Thus, using anti-CD4 CAR-NK/T cells in AITL patients might represent a promising strategy, even though healthy CD4 T cells will also be depleted. This latter means that in the long term this approach can lead to CD4 T-cell elimination and immunosuppression. If depleting normal B cells seem to be well-tolerated in B-cell malignancies by CAR therapies, there is no sufficient insight to extrapolate this to T-cell lymphomas yet. Pinz et al.
[61][97] constructed anti-CD4 CAR-NK that specifically eliminated robustly diverse ex vivo CD4+ human T-cell leukemia and lymphoma cell lines in vivo. These preclinical results are encouraging for anti-CD4 CAR-NK therapy use in case of all CD4+ T-cell malignancies and in particular for AITL
[61][97]. In fact, clinical trials are ongoing using anti-CD4 CAR-T cells for relapsed/refractory T-cell lymphoma, including AITL (NCT04712864). The advantage of an anti-CD4 NK-cell application might be that NK cells are short lived in vivo compared to CAR-T cells and do not lead to extended healthy CD4 T-cell immunosuppression in the patients. However, this still needs to be consolidated.
While targeting the CD4 antigen also depletes normal CD4 T cells, M. Maciocia et al.
[62][98] used a more specific approach to target malignant T cells. The TCR comprises a heterodimeric protein complex of two chains, TCRα and TCRβ. An ancestral duplication of the β-chain constant gene results in the expression of one of two highly homologous chains, T-cell receptor β-chain constant (TRBC) domains 1 and 2, in a mutually exclusive manner following the TCR locus rearrangement. Based on the mutually exclusive expression of TRBC1 and TRBC2, they developed anti-TRBC1 CAR-T cells that recognized and killed normal and malignant TRBC1+ but not TRBC2+ T cells in vitro and in a disseminated mouse model of leukemia. This strategy allows the selective targeting and depletion of T cells carrying the TRBC1 chain, both healthy and malignant, while sparing healthy T cells expressing TRBC2, thereby preserving T-cell-mediated immune responses
[62][98]. A phase I/II study at the recruiting phase will evaluate AUTO4 (anti-TRBC1 CAR-T cells) in AITL patients with TRBC1 CD4+ malignant cell clonality (NCT03590574 and NCT0482817).
Finally, as previously described, CD30 is a tumor cell marker in AITL. To explore the CD30 potency as a target, clinical trials at the recruiting stage are ongoing to explore anti-CD30 CAR-T cells as a therapy to treat AITL patients and other T-cell lymphomas and leukemia (NCT04008394).