Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | David Yang | -- | 1708 | 2022-05-23 02:13:00 | | | |
| 2 | David Yang | + 3 word(s) | 1711 | 2022-05-23 02:17:49 | | | | |
| 3 | Vivi Li | -49 word(s) | 1662 | 2022-05-23 04:07:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yang, D.; Ta, R.; , .; Flavin, R. Diffuse Large B-Cell Lymphoma and Its Tumor Microenvironment. Encyclopedia. Available online: https://encyclopedia.pub/entry/23219 (accessed on 09 August 2026).
Yang D, Ta R, , Flavin R. Diffuse Large B-Cell Lymphoma and Its Tumor Microenvironment. Encyclopedia. Available at: https://encyclopedia.pub/entry/23219. Accessed August 09, 2026.
Yang, David, Robert Ta, , Richard Flavin. "Diffuse Large B-Cell Lymphoma and Its Tumor Microenvironment" Encyclopedia, https://encyclopedia.pub/entry/23219 (accessed August 09, 2026).
Yang, D., Ta, R., , ., & Flavin, R. (2022, May 23). Diffuse Large B-Cell Lymphoma and Its Tumor Microenvironment. In Encyclopedia. https://encyclopedia.pub/entry/23219
Yang, David, et al. "Diffuse Large B-Cell Lymphoma and Its Tumor Microenvironment." Encyclopedia. Web. 23 May, 2022.
Copy Citation
Diffuse large B-cell lymphoma (DLBCL) is the most common non-Hodgkin lymphoma. It is a clinically and morphologically heterogeneous entity that has continued to resist complete subtyping. Molecular subtyping efforts emerged in earnest with the advent of gene expression profiling (GEP). This molecular subtyping approach has continued to evolve simultaneously with others including immunohistochemistry and more modern genomic approaches. The veritable explosion of genomic data availability and evolving computational methodologies have provided additional avenues, by which further understanding and subclassification of DBLCLs is possible.
molecular
diagnostics
tumor microenvironment
diffuse large B-cell lymphoma
cell-of-origin
omics
immunohistochemistry
next generation sequencing
gene expression profiling
cytogenetics/FISH
minimum criteria for diagnosis
1. Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common non-Hodgkin lymphoma in the western hemisphere [1]. Diagnosis and subtyping of DLBCL has come far to date, from just requiring morphological assessment based on a single H & E slide to now, where numerous ancillary tests are a prerequisite, including immunohistochemistry, cytogenetics, flow cytometry, and molecular testing. Whilst these technologies continue to allow refinements in diagnostic subtyping, the highly heterogeneous nature of the disease continues to evade full subtyping efforts. Furthermore, continued efforts to understand the underlying molecular pathophysiology of the disease are needed, given the propensity of the disease to relapse beyond standard and even precision therapies.
2. Gene Expression Profiling
It was not long ago that the diagnosis of liquid tumors rested on morphology, immunohistochemical analyses, and cytogenetics. The original diffuse large B-cell lymphoma (DLBCL) molecular subclassification followed the advent of DNA microarrays, a technology that allows the analysis of thousands of expressed genes simultaneously; see Figure 1 for a theoretical example [2]. This DNA microarray-based technology allowed for transcriptional gene pattern expression (e.g., Lymphochip by Alizadeh et al. [3]) analysis under defined conditions that delineated the seminal molecular study classifying a liquid cancer DLBCL into the so-called cell of origin (COO) subtypes [4]. This accomplishment resulted in the subtyping of approximately 80–85% of all DLBCL cases and importantly showed subtype prognostic values that were greater than that of the standard clinical predictor, the International Prognostic Index (IPI) [4]. In addition, this critical work paved the way for immunohistochemical (IHC) determination of COO.
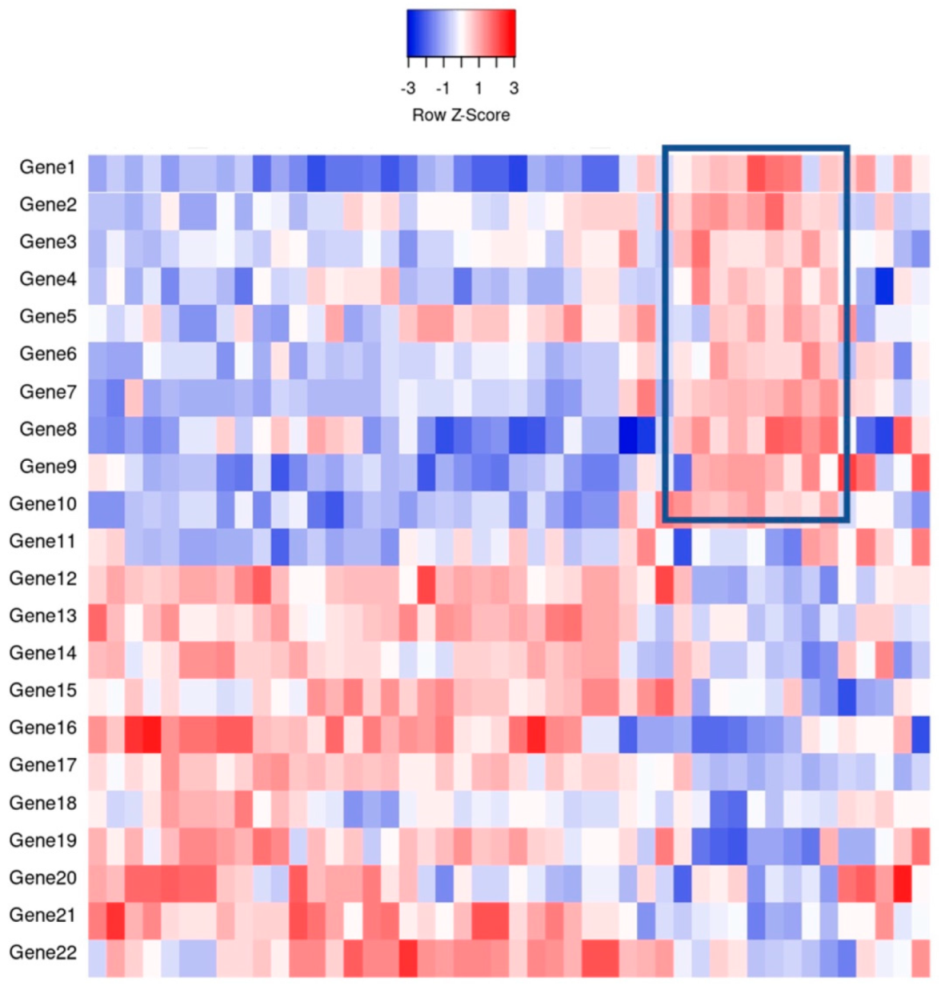
Figure 1. An example of gene expression profiling comparing the relative gene expression levels and grouping of cases by clusters. For example, the indicated boxed area indicates that Genes 1–10 have a relative increase in expression, thereby potentially clustering cases together for classification purposes.
Although the study population was small by current standards, with imperfect subtyping, the classification was adopted and continues today as the standard of diagnostic care, recognized within the World Health Organization’s Revised Fourth Edition of the Classification of Tumors of the Hematopoietic and Lymphoid Tissues [5] as germinal center B-cell like (GCB) and activated B-cell like (ABC), with the remainder left as unclassified. Novelty is never without controversy as other approaches, including an unbiased a priori approach which used supervised machine learning to analyze GEP, did not find molecular correlates of COO to be independently prognostic [6]. Nevertheless, the same group reported differing GEP signatures that predicted response to CHOP chemotherapy (cyclophosphamide, doxorubicin hydrochloride, vincristine, and prednisone). Indeed, in that vein, groups like Rosenwald et al. [7] reported cell specific and non-cell specific genetic signatures that differed depending on response to standard chemotherapies. Regardless of molecular prognostication and subtyping, molecular investigation was certain to provide increasing identification of precision therapeutic targets based on biochemical pathways [4][6][7][8].
Despite the reasonable success of standard CHOP and R-CHOP, and the growing number of precision therapies, heterogeneity remained an issue, as evidenced by inconsistent treatment responses and relapses despite COO subtyping. Although gaining favor and resolving power, it was thought perhaps that the transcriptionally based GEP was an inadequate representation of underlying aberrant genetic programming (e.g., a DNA repair protein with single nucleotide polymorphism). However, practically, other technical hurdles, such as the logistical necessity for fresh tissue specimens, stood in the way of immediate clinical adoption of GEP in the subcategorization of DLBCLs. Initially, this was a difficult step to overcome, however several novel testing options were eventually able to surmount this issue, including Nanostring [9], HTG [10], and Roche [11]. These options allowed the use of formalin fixed paraffin embedded (FFPE) tissues, which was in keeping with standard pathologic workflow and also allowed for an increase in analytical case numbers to be studied.
Meanwhile, through whole exome sequencing and transcriptome sequencing, oncogenic drivers of DLBCL were mapped [12]. These findings reported that the most unfavorable prognoses were DLBCL cases with MYC aberrations along with MYC IHC over-expression. Other studies using mouse modelling also supported the molecular ideas emerging from GEP, showing oncogenic driver mutations in genes such as EZH2 and MYD88 that promoted lymphoma development [13][14]. Simultaneously on the molecular subclassification side, other analytic hurdles, such as setup and integration of the multiple platforms (e.g., whole-exome sequencing and RNA sequencing), were required to adequately identify different types of genetic aberrations, including mutations, translocations, and copy number alterations [15][16].
Through previously mentioned genomic methods, Chapuy et al. [15] utilized clustering analytic methods to identify low-frequency alterations, captured recurrent mutations, somatic copy number alterations, and structural variants, and created five differential genetic signatures to further subtype DLBCL (Figure 2). C1 was ABC-related, associated with NOTCH2 mutations and favorable outcome; C2 was unrelated to GCB and ABC, had frequent biallelic TP53 inactivation, CDKN2A deletion, and poor outcomes; C3 was GCB-related, associated with BCL2 translocation, PTEN aberrations, epigenetic modifiers (KMT2D, CREBBP, and EZH2) and unfavorable outcome; C4 was GCB-related, with BCR–PI3K, NF-κB, or RAS–JAK signal transducer, was an activator of transcription (BRAF and STAT3) pathway aberrations, histone gene mutations, cluster of differentiation proteins associated with immune evasion (CD83, CD70, and CD58), and had favorable outcomes; C5 was ABC related, gained BCL2, MYD88L265P, CD79B, PIM1, and PRDM1 mutations, and had unfavorable outcome. The prognostic capability of the subtypes was also independent of the clinical gold standard IPI [15].
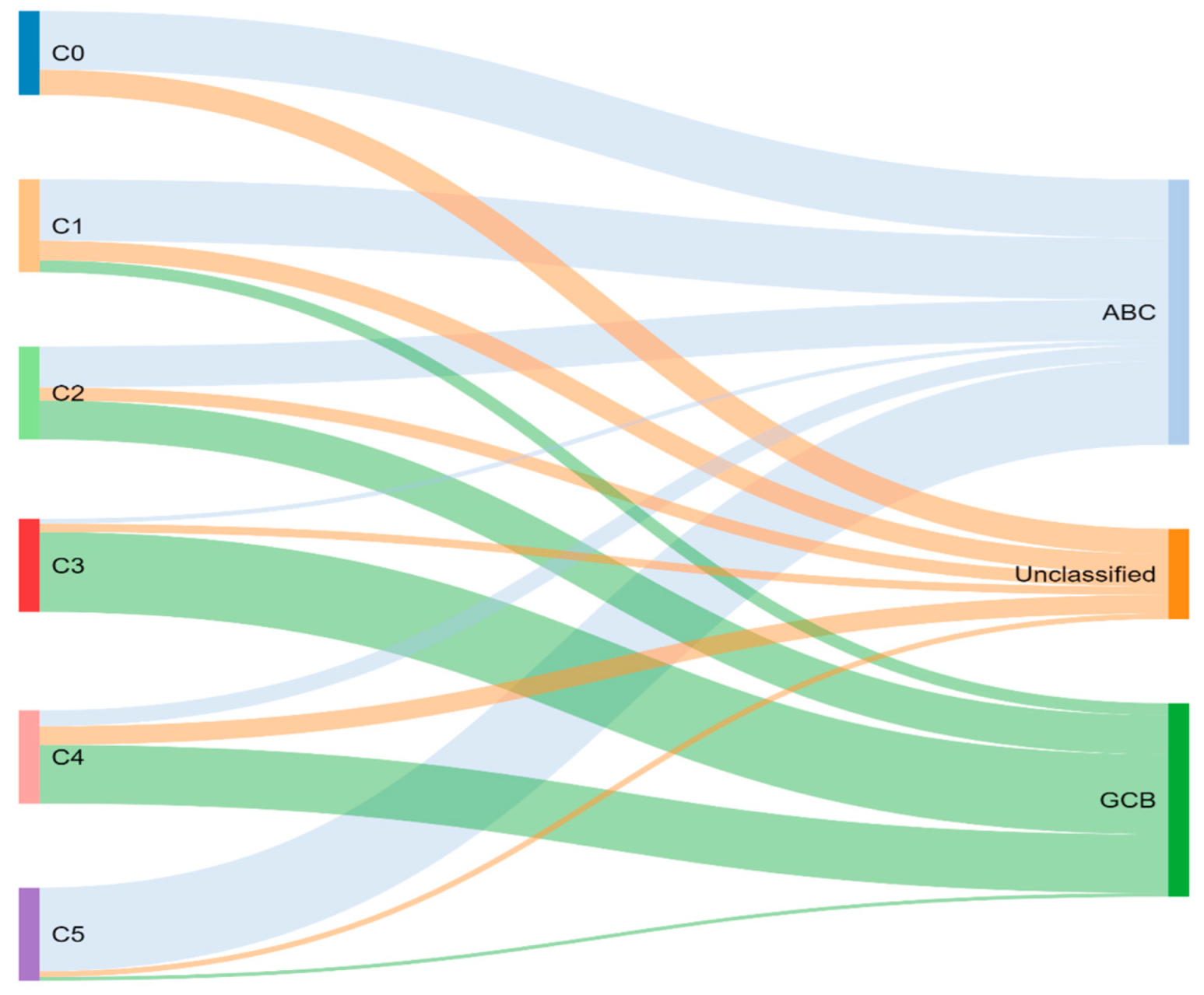
Figure 2. Chapuy and colleagues’ genetic cluster classification of DLBCL subtype, as compared to cell of origin. C0–C5 = cluster 0 to cluster 5.
Concurrently, Schmitz et al. [16] simultaneously characterized four DLBCL subsets via their GenClass algorithm, termed MCD (MYD88L265P, CD79B co-mutation), BN2 (BCL6 fusions or NOTCH2 mutation), N1 (NOTCH1 mutations), or EZB (EZH2 mutation or BCL2 translocation), based on a more homogenous set of genomic aberrations (Figure 3). Interestingly, they were able to identify precision targets within the high-risk subtypes, showing, for example, that MCD could be more responsive to ibrutinib secondary to constitutive BCR signaling [16].
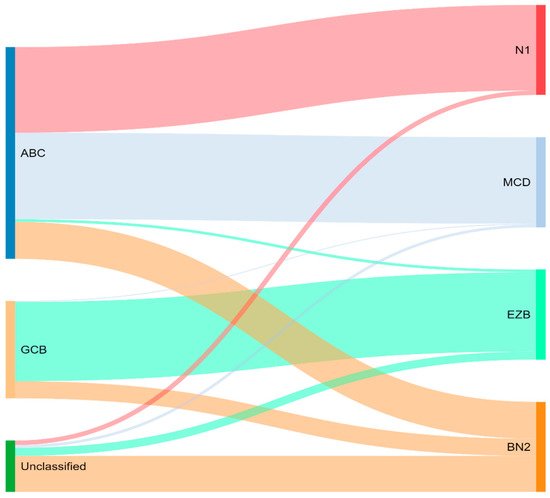
Figure 3. Schmitz and colleagues’ genetic classification of DLBCL subtype as compared to cell of origin. ABC = activated B-cell, GCB = germinal-center B-cell, N1 = notch 1, MCD = MYD88L265P, CD79B co-mutation, EZB = EZH2 mutation or BCL2 translocation, and BN2 = Notch 2.
To compare, the Chapuy C1, C3, and C5 clusters overlapped with the Schmitz GenClass BN2, EZB, and MCD groups, respectively. The Chapuy C2 and C4 subtypings did not overlap with any of the other Schmitz subtypings. This non-concordance was perhaps thought secondary to differences in bioinformatic analytic approaches.
For the last two decades, R-CHOP has been the standard of treatment in previously untreated DLBCL. With the molecular subtyping of DLBCL, it was proposed that a subtype specific treatment could improve response rates specially for patients who do not achieve complete remission or develop disease relapse (around 40% treated with R-CHOP) [17]. Ibrutinib, a first-in-class oral covalent inhibitor of Bruton’s tyrosine kinase (BTK) showed some preferential activity in ABC DLBCL [18]. In a randomized multicenter study [19], the goal was to determine if addition of ibrutinib would improve efficacy of R-CHOP in ABC DLBCL. Interestingly, the addition of ibrutinib to R-CHOP improved event-free survival and overall survival in patients younger than 60 years. Unfortunately, older patients (>60 years of age) had increased serious adverse effects with ibrutinib plus R-CHOP. Of note, molecular subtyping increased median time to diagnosis by 27 days, which may have excluded patients necessitating immediate treatment. The study shows the potential of subtype specific treatment as well the need for reasonable turnaround diagnostic times if integrating molecular subtyping. Subsequently, the PHOENIX trial continued to demonstrate superior outcomes in younger patients treated with ibrutinib and R-CHOP, but also better overall survival in specific molecular subsets including the MCD and N1 subgroups compared to R-CHOP alone [20]. Landsburg and colleagues found that ibrutinib monotherapy had a 60% response rate in relapsed/refractory patients with a non-germinal center, and MYC and BCL2 double expressor phenotype [21].
The ROBUST study is a phase 3 clinical trial that compared the addition of lenalidomide to rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) therapy with R-CHOP therapy alone for treatment of activated B-cell-like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL). ABC-type DLBCL has traditionally been shown to resist typical R-CHOP therapy, however, emerging phase 2 studies are demonstrating promise of the addition of lenalidomide to R-CHOP (R2-CHOP) in ABC-type therapy. The primary end point of the study was progression-free survival (PFS) of participants receiving R2-CHOP, compared to those receiving R-CHOP only. Although PFS was not met (hazard ratio 0.85), the median PFS was not reached for either group. PFS tended to favor R2-CHOP over placebo group in patients with higher-risk disease, but adverse events of R2-CHOP compared to placebo were neutropenia (60% vs. 48%), anemia (22% vs. 14%), thrombocytopenia (17% vs. 11%), and leukopenia (14% vs. 15%). Of note, ROBUST was the first phase 3 study to highlight biomarker identification of ABC patients and was able to demonstrate a consistent safety profile of R2-CHOP.
Initial hopes were high that the development of molecular GEP would have significant effects on prognostic DLBCL classification, leading to therapeutic tailoring. With the advent of GEP, studies naturally attempted retrospective gene expression profiling analyses on their DLBCL cohorts, unfortunately, with conflicting results. Davies et al. (REMoDL-B) were the first to show that GEP for therapeutic assignment was possible prospectively [22]. However, their randomized phase 3 clinical trial results were disappointing, reporting that the addition of bortezomib to suppress the GEP apparently increased NF-kB gene expression to standard R-CHOP, and failed to improve survival in ABC DLBCLs. While the trial itself was not without criticism, the study lent some doubt as to whether GEP classification was a prognostic and therapeutic breakthrough…
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression Monitoring. Science 1999, 286, 531–537.
- Alizadeh, A.A.; Eisen, M.; Davis, R.; Ma, C.; Sabet, H.; Tran, T.; Powell, J.I.; Yang, L.; Marti, G.; Moore, D.T.; et al. The Lymphochip: A Specialized cDNA Microarray for the Genomic-scale Analysis of Gene Expression in Normal and Malignant Lymphocytes. Cold Spring Harb. Symp. Quant. Biol. 1999, 64, 71–78.
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. (Eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; IARC: Lyon, France, 2017.
- Shipp, M.A.; Ross, K.N.; Tamayo, P.; Weng, A.; Kutok, J.L.; Aguiar, R.C.; Gaasenbeek, M.; Angelo, M.; Reich, M.; Pinkus, G.S.; et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat. Med. 2002, 8, 68–74.
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The Use of Molecular Profiling to Predict Survival after Chemotherapy for Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947.
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal Gene Signatures in Large-B-Cell Lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323.
- Scott, D.W.; Wright, G.W.; Williams, P.M.; Lih, C.-J.; Walsh, W.; Jaffe, E.; Rosenwald, A.; Campo, E.; Chan, W.C.; Connors, J.M.; et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 2014, 123, 1214–1217.
- Balasubramanian, S.; Wang, S.; Major, C.; Hodkinson, B.; Schaffer, M.; Sehn, L.H.; Johnson, P.; Zinzani, P.L.; Carey, J.; Shreeve, S.M.; et al. Comparison of immunohistochemistry and gene expression profiling subtyping for diffuse large B-cell lymphoma in the phase III clinical trial of R-CHOP ± ibrutinib. Br. J. Haematol. 2021, 194, 83–91.
- Ta, R.; Santini, C.; Gou, P.; Lee, G.; Tai, Y.C.; O’Brien, C.; Fontecha, M.; Grant, C.; Bacon, L.; Finn, S.; et al. Molecular Subtyping of Diffuse Large B-Cell Lymphoma Using a Novel Quantitative RT-PCR Assay. J. Mol. Diagn. 2020, 23, 323–340.
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.; Love, C.L.; Waldrop, A.; Leppä, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15.
- Knittel, G.; Liedgens, P.; Korovkina, D.; Seeger, J.M.; Al-Baldawi, Y.; Al-Maarri, M.; Fritz, C.; Vlantis, K.; Bezhanova, S.; Scheel, A.H.; et al. B-cell–specific conditional expression of Myd88p.L252P leads to the development of diffuse large B-cell lymphoma in mice. Blood 2016, 127, 2732–2741.
- Souroullas, G.P.; Jeck, W.R.; Parker, J.S.; Simon, J.M.; Liu, J.-Y.; Paulk, J.; Xiong, J.; Clark, K.S.; Fedoriw, Y.; Qi, J.; et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 2016, 22, 632–640.
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690.
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407.
- Nowakowski, G.S.; Chiappella, A.; Witzig, T.E.; Spina, M.; Gascoyne, R.D.; Zhang, L.; Flament, J.; Repici, J.; Vitolo, U. ROBUST: Lenalidomide-R-CHOP versus placebo-R-CHOP in previously untreated ABC-type diffuse large B-cell lymphoma. Futur. Oncol. 2016, 12, 1553–1563.
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.-J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926.
- Younes, A.; Sehn, L.H.; Johnson, P.; Zinzani, P.L.; Hong, X.; Zhu, J.; Patti, C.; Belada, D.; Samoilova, O.; Suh, C.; et al. Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 1285–1295.
- Wilson, W.H.; Wright, G.W.; Huang, D.W.; Hodkinson, B.; Balasubramanian, S.; Fan, Y.; Vermeulen, J.; Shreeve, M.; Staudt, L.M. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell 2021, 39, 1643–1653.e3.
- Landsburg, D.J.; Hughes, M.E.; Koike, A.; Bond, D.; Maddocks, K.J.; Guo, L.; Winter, A.M.; Hill, B.T.; Ondrejka, S.L.; Hsi, E.D.; et al. Outcomes of patients with relapsed/refractory double-expressor B-cell lymphoma treated with ibrutinib monotherapy. Blood Adv. 2019, 3, 132–135.
- Davies, A.; Cummin, T.E.; Barrans, S.; Maishman, T.; Mamot, C.; Novak, U.; Caddy, J.; Stanton, L.; Kazmi-Stokes, S.; McMillan, A.; et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): An open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 649–662.
More
Information
Subjects:
Hematology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
3 times
(View History)
Update Date:
23 May 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No