 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zhijun Luo | -- | 1907 | 2022-05-19 06:30:54 | | | |
| 2 | Beatrix Zheng | Meta information modification | 1907 | 2022-05-19 08:56:23 | | |
Video Upload Options
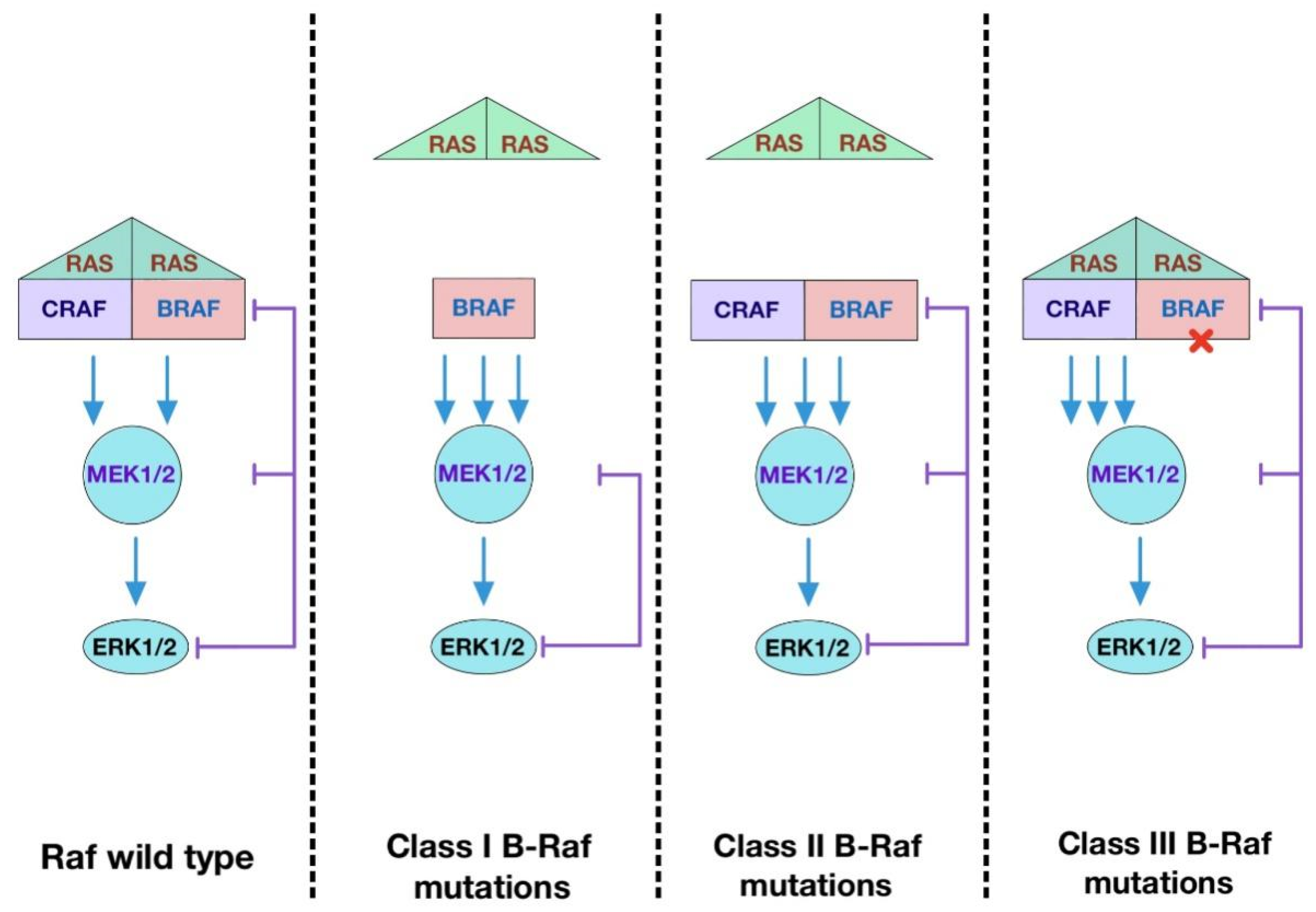
The Raf kinase family consists of three isoforms, C-Raf/Raf-1, B-Raf and A-Raf. They are located directly downstream of Ras and upstream of MEK1/2. Since the discovery of v-Ras, v-Raf and ERK and the connection of this regulatory hub with oncogenesis, tremendous efforts have been invested in the elucidation of the mechanisms underlying the activation of Raf kinases. The Raf/MEK/ERK pathway is the first and clearly defined mitogenic pathway whose signal is invoked by extracellular mitogenic ligands and serves as a framework for other MAPK pathways. Among three isoforms of the Raf family, B-Raf is the only one that has so far been found mutated in many types of cancers. As all of the Raf family members directly act downstream of Ras, C-Raf and A-Raf are also important factors contributing to oncogenesis, either mediating the effects of mutated Ras or participating in oncogenic B-Raf-mediated pathogenesis. Thus, the development of Raf inhibitors has been a focus in cancer therapy.
1. Introduction
2. Discovery of the Raf/MEK/ERK Pathway
3. Role of Raf in Biology
References
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell. Biol. 2015, 16, 281–298.
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260.
- Kyriakis, J.M.; App, H.; Zhang, X.F.; Banerjee, P.; Brautigan, D.L.; Rapp, U.R.; Avruch, J. Raf-1 activates MAP kinase-kinase. Nature 1992, 358, 417–421.
- Zhang, X.F.; Settleman, J.; Kyriakis, J.M.; Takeuchi-Suzuki, E.; Elledge, S.J.; Marshall, M.S.; Bruder, J.T.; Rapp, U.R.; Avruch, J. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 1993, 364, 308–313.
- Rapp, U.R.; Goldsborough, M.D.; Mark, G.E.; Bonner, T.I.; Groffen, J.; Reynolds, F.H., Jr.; Stephenson, J.R. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc. Natl. Acad. Sci. USA 1983, 80, 4218–4222.
- Brown, K.; Quintanilla, M.; Ramsden, M.; Kerr, I.B.; Young, S.; Balmain, A. v-ras genes from Harvey and BALB murine sarcoma viruses can act as initiators of two-stage mouse skin carcinogenesis. Cell 1986, 46, 447–456.
- Shimizu, K.; Goldfarb, M.; Suard, Y.; Perucho, M.; Li, Y.; Kamata, T.; Feramisco, J.; Stavnezer, E.; Fogh, J.; Wigler, M.H. Three human transforming genes are related to the viral ras oncogenes. Proc. Natl. Acad. Sci. USA 1983, 80, 2112–2116.
- Ladeda, V.; Frankel, P.; Feig, L.A.; Foster, D.A.; Bal de Kier Joffe, E.; Aguirre-Ghiso, J.A. RalA mediates v-Src, v-Ras, and v-Raf regulation of CD44 and fibronectin expression in NIH3T3 fibroblasts. Biochem. Biophys. Res. Commun. 2001, 283, 854–861.
- Pisapia, P.; Pepe, F.; Iaccarino, A.; Sgariglia, R.; Nacchio, M.; Russo, G.; Gragnano, G.; Malapelle, U.; Troncone, G. BRAF: A Two-Faced Janus. Cells 2020, 9, 2549.
- Leevers, S.J.; Marshall, C.J. Activation of extracellular signal-regulated kinase, ERK2, by p21ras oncoprotein. EMBO J. 1992, 11, 569–574.
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27.
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465.
- Ahn, N.G. The MAP kinase cascade. Discovery of a new signal transduction pathway. Mol. Cell. Biochem. 1993, 127–128, 201–209.
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Kwong, L.N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696.
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691.
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell. Biol. 2003, 4, 373–384.
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319.
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457.
- Rajagopalan, H.; Bardelli, A.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002, 418, 934.
- Never-smoker, N.E.S. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550.
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315.
- Rapp, U.R.; Reynolds, F.H., Jr.; Stephenson, J.R. New mammalian transforming retrovirus: Demonstration of a polyprotein gene product. J. Virol. 1983, 45, 914–924.
- Rapp, U.R.; Cleveland, J.L.; Fredrickson, T.N.; Holmes, K.L.; Morse, H.C., 3rd; Jansen, H.W.; Patschinsky, T.; Bister, K. Rapid induction of hemopoietic neoplasms in newborn mice by a raf(mil)/myc recombinant murine retrovirus. J. Virol. 1985, 55, 23–33.
- Jansen, H.W.; Lurz, R.; Bister, K.; Bonner, T.I.; Mark, G.E.; Rapp, U.R. Homologous cell-derived oncogenes in avian carcinoma virus MH2 and murine sarcoma virus 3611. Nature 1984, 307, 281–284.
- Rapp, U.R.; Todaro, C. Generation of new mouse sarcoma viruses in cell culture. Science 1978, 201, 821–824.
- Moelling, K.; Heimann, B.; Beimling, P.; Rapp, U.R.; Sander, T. Serine- and threonine-specific protein kinase activities of purified gag-mil and gag-raf proteins. Nature 1984, 312, 558–561.
- Bonner, T.I.; Kerby, S.B.; Sutrave, P.; Gunnell, M.A.; Mark, G.; Rapp, U.R. Structure and biological activity of human homologs of the raf/mil oncogene. Mol. Cell. Biol. 1985, 5, 1400–1407.
- Bonner, T.; O’Brien, S.J.; Nash, W.G.; Rapp, U.R.; Morton, C.C.; Leder, P. The human homologs of the raf (mil) oncogene are located on human chromosomes 3 and 4. Science 1984, 223, 71–74.
- Ikawa, S.; Fukui, M.; Ueyama, Y.; Tamaoki, N.; Yamamoto, T.; Toyoshima, K. B-raf, a new member of the raf family, is activated by DNA rearrangement. Mol. Cell. Biol. 1988, 8, 2651–2654.
- Huleihel, M.; Goldsborough, M.; Cleveland, J.; Gunnell, M.; Bonner, T.; Rapp, U.R. Characterization of murine A-raf, a new oncogene related to the v-raf oncogene. Mol. Cell. Biol. 1986, 6, 2655–2662.
- Han, M.; Golden, A.; Han, Y.; Sternberg, P.W.C. C. elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature 1993, 363, 133–140.
- Ambrosio, L.; Mahowald, A.P.; Perrimon, N. Requirement of the Drosophila raf homologue for torso function. Nature 1989, 342, 288–291.
- Barnier, J.V.; Papin, C.; Eychène, A.; Lecoq, O.; Calothy, G. The mouse B-raf gene encodes multiple protein isoforms with tissue-specific expression. J. Biol. Chem. 1995, 270, 23381–23389.
- Storm, S.M.; Cleveland, J.L.; Rapp, U.R. Expression of raf family proto-oncogenes in normal mouse tissues. Oncogene 1990, 5, 345–351.
- Sturgill, T.W.; Ray, L.B.; Erikson, E.; Maller, J.L. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature 1988, 334, 715–718.
- Courchesne, W.E.; Kunisawa, R.; Thorner, J. A putative protein kinase overcomes pheromone-induced arrest of cell cycling in S. cerevisiae. Cell 1989, 58, 1107–1119.
- Ray, L.B.; Sturgill, T.W. Insulin-stimulated microtubule-associated protein kinase is phosphorylated on tyrosine and threonine in vivo. Proc. Natl. Acad. Sci. USA 1988, 85, 3753–3757.
- Lindberg, R.A.; Quinn, A.M.; Hunter, T. Dual-specificity protein kinases: Will any hydroxyl do? Trends Biochem. Sci. 1992, 17, 114–119.
- Anderson, N.G.; Maller, J.L.; Tonks, N.K.; Sturgill, T.W. Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature 1990, 343, 651–653.
- Ullah, R.; Yin, Q.; Snell, A.H.; Wan, L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin. Cancer Biol. 2021.
- Dent, P.; Haser, W.; Haystead, T.A.; Vincent, L.A.; Roberts, T.M.; Sturgill, T.W. Activation of mitogen-activated protein kinase kinase by v-Raf in NIH 3T3 cells and in vitro. Science 1992, 257, 1404–1407.
- Gómez, N.; Cohen, P. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature 1991, 353, 170–173.
- Ahn, N.G.; Seger, R.; Bratlien, R.L.; Diltz, C.D.; Tonks, N.K.; Krebs, E.G. Multiple components in an epidermal growth factor-stimulated protein kinase cascade. In vitro activation of a myelin basic protein/microtubule-associated protein 2 kinase. J. Biol. Chem. 1991, 266, 4220–4227.
- Crews, C.M.; Alessandrini, A.; Erikson, R.L. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science 1992, 258, 478–480.
- Zheng, C.F.; Guan, K.L. Cloning and characterization of two distinct human extracellular signal-regulated kinase activator kinases, MEK1 and MEK2. J. Biol. Chem. 1993, 268, 11435–11439.
- Vojtek, A.B.; Hollenberg, S.M.; Cooper, J.A. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 1993, 74, 205–214.
- Warne, P.H.; Viciana, P.R.; Downward, J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993, 364, 352–355.
- Pritchard, C.A.; Bolin, L.; Slattery, R.; Murray, R.; McMahon, M. Post-natal lethality and neurological and gastrointestinal defects in mice with targeted disruption of the A-Raf protein kinase gene. Curr. Biol. 1996, 6, 614–617.
- Wojnowski, L.; Zimmer, A.M.; Beck, T.W.; Hahn, H.; Bernal, R.; Rapp, U.R.; Zimmer, A. Endothelial apoptosis in Braf-deficient mice. Nat. Genet. 1997, 16, 293–297.
- Wojnowski, L.; Stancato, L.F.; Zimmer, A.M.; Hahn, H.; Beck, T.W.; Larner, A.C.; Rapp, U.R.; Zimmer, A. Craf-1 protein kinase is essential for mouse development. Mech. Dev. 1998, 76, 141–149.
- Wiese, S.; Pei, G.; Karch, C.; Troppmair, J.; Holtmann, B.; Rapp, U.R.; Sendtner, M. Specific function of B-Raf in mediating survival of embryonic motoneurons and sensory neurons. Nat. Neurosci. 2001, 4, 137–142.
- De Iriarte Rodríguez, R.; Magariños, M.; Pfeiffer, V.; Rapp, U.R.; Varela-Nieto, I. C-Raf deficiency leads to hearing loss and increased noise susceptibility. Cell Mol. Life Sci. 2015, 72, 3983–3998.
- Hüser, M.; Luckett, J.; Chiloeches, A.; Mercer, K.; Iwobi, M.; Giblett, S.; Sun, X.M.; Brown, J.; Marais, R.; Pritchard, C. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 2001, 20, 1940–1951.
- Mikula, M.; Schreiber, M.; Husak, Z.; Kucerova, L.; Rüth, J.; Wieser, R.; Zatloukal, K.; Beug, H.; Wagner, E.F.; Baccarini, M. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J. 2001, 20, 1952–1962.
- Rebocho, A.P.; Marais, R. ARAF acts as a scaffold to stabilize BRAF:CRAF heterodimers. Oncogene 2013, 32, 3207–3212.
- Dumaz, N.; Hayward, R.; Martin, J.; Ogilvie, L.; Hedley, D.; Curtin, J.A.; Bastian, B.C.; Springer, C.; Marais, R. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006, 66, 9483–9491.
- McPhillips, F.; Mullen, P.; MacLeod, K.G.; Sewell, J.M.; Monia, B.P.; Cameron, D.A.; Smyth, J.F.; Langdon, S.P. Raf-1 is the predominant Raf isoform that mediates growth factor-stimulated growth in ovarian cancer cells. Carcinogenesis 2006, 27, 729–739.
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954.
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867.
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell. Biol. 2004, 5, 875–885.
- Kumar, R.; Angelini, S.; Czene, K.; Sauroja, I.; Hahka-Kemppinen, M.; Pyrhönen, S.; Hemminki, K. BRAF mutations in metastatic melanoma: A possible association with clinical outcome. Clin. Cancer Res. 2003, 9, 3362–3368.
- Dhomen, N.; Marais, R. New insight into BRAF mutations in cancer. Curr. Opin. Genet. Dev. 2007, 17, 31–39.
- Garnett, M.J.; Rana, S.; Paterson, H.; Barford, D.; Marais, R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol. Cell 2005, 20, 963–969.
- Tran, N.H.; Frost, J.A. Phosphorylation of Raf-1 by p21-activated kinase 1 and Src regulates Raf-1 autoinhibition. J. Biol. Chem. 2003, 278, 11221–11226.
- Zebisch, A.; Staber, P.B.; Delavar, A.; Bodner, C.; Hiden, K.; Fischereder, K.; Janakiraman, M.; Linkesch, W.; Auner, H.W.; Emberger, W.; et al. Two transforming C-RAF germ-line mutations identified in patients with therapy-related acute myeloid leukemia. Cancer Res. 2006, 66, 3401–3408.
- Pandit, B.; Sarkozy, A.; Pennacchio, L.A.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 2007, 39, 1007–1012.
- Razzaque, M.A.; Nishizawa, T.; Komoike, Y.; Yagi, H.; Furutani, M.; Amo, R.; Kamisago, M.; Momma, K.; Katayama, H.; Nakagawa, M.; et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat. Genet. 2007, 39, 1013–1017.
- Rauen, K.A. The RASopathies. Annu. Rev. Genomics Hum. Genet. 2013, 14, 355–369.
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383.
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238.
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341.

