1. Introduction
The Raf kinase family consists of three isoforms, C-Raf/Raf-1, B-Raf and A-Raf
[1][2][1,2]. They are located directly downstream of Ras and upstream of MEK1/2
[3][4][3,4]. Since the discovery of v-Ras, v-Raf and ERK
[5][6][7][8][9][10][11][12][5,6,7,8,9,10,11,12] and the connection of this regulatory hub with oncogenesis, tremendous efforts have been invested in the elucidation of the mechanisms underlying the activation of Raf kinases. The Raf/MEK/ERK pathway is the first and clearly defined mitogenic pathway whose signal is invoked by extracellular mitogenic ligands and serves as a framework for other MAPK pathways
[13]. Among three isoforms of the Raf family,
B-Raf is the only one that has so far been found mutated in many types of cancers
[14]. As all of the Raf family members directly act downstream of Ras, C-Raf and A-Raf are also important factors contributing to oncogenesis, either mediating the effects of mutated
Ras or participating in oncogenic
B-Raf-mediated pathogenesis. Thus, the development of Raf inhibitors has been a focus in cancer therapy.
The Raf/MEK/ERK pathway plays important roles not only in physiological processes, including cell proliferation, differentiation and development, but also in oncogenesis and cancer progression
[15]. The oncogenic
Ras isoforms,
KRas,
HRas and
NRas, whose mutations have been found in more than 30% of human cancers, as well as overexpression of growth factors and mutations of their receptors in human cancer, all lead to the activation of this pathway
[11][12][16][11,12,16]. Furthermore,
B-Raf mutations are present in approximately 8% of human cancers
[17], including 50% of melanoma
[14], 45% of papillary thyroid cancer
[18], 10% of colon cancer
[19], 10% of non-small cell lung cancer
[20] and almost 100% of hairy cell leukemia cases
[21]. Thus, the development of drugs targeting the Raf/MEK/ERK pathway is especially important for cancer therapy. To fulfill this objective, the elucidation of this pathway is a crucial step. Although the mechanisms of MEK and ERK activation are relatively straightforward, Raf activation is rather complex and still incompletely understood. Moreover, the discovery of homo- and hetero-dimerization of Raf isoforms adds to the complexity of deciphering the mechanism of their activation, which also makes the development of Raf inhibitors a challenging and daunting task. Therefore, this re
svie
archw will summarize current knowledge of the regulation of Raf isoforms and progress in the drug development of Raf inhibitors for cancer therapy.
2. Discovery of the Raf/MEK/ERK Pathway
In the early 1980s,
v-Raf was initially identified as a transforming gene of the murine retrovirus 3611-murine sarcoma virus (MSV)
[5][22][5,22]. In neonatal mice, it causes predominantly fibrosarcoma and erythroleukemias. Hence, the name of Raf originated from its capability to stimulate “Rapidly Accelerated
Fibrosarcomas”
[23]. Shortly after, the genome of the avian carcinoma virus MH2 was found to encode a closely related oncogene named
v-mil [24]. Comparison of DNA sequences of these oncogenes coupled with biochemical studies has revealed that
v-Raf and
v-mil are retroviral oncogenes derived from cellular proto-oncogenes of mammalian and avian species. Both genes encode products that are classified into the serine/threonine kinase family, homologous to tyrosine specific SRC kinases in the kinase domain.
v-Raf and
v-mil are fused to the N-myristoylated (N-myr) viral Gag sequence but with the deletion of amino-terminal moieties, in contrast to their cellular counterparts
[25][26][25,26].
The first cellular counterparts of
v-Raf,
C-Raf-1 and
C-Raf-2, were cloned and sequenced in 1985. However, it soon became apparent that
C-Raf-2 is a pseudogene. Thus, the
C-Raf-1 gene product was designated as C-Raf. Studies have reported that
C-Raf is located at human chromosomal band 3p25, consisting of nine exons that are similar to v-
Raf and v-
mil as well as two extra exons that are related to v-
mil [27][28][27,28]. Later on, two more Raf family members, A-Raf and B-Raf, were identified in vertebrates
[2][29][30][2,29,30]. Although C-Raf was the first to be discovered in mammals, the ortholog of B-Raf is D-Raf in
Drosophila melanogaster and LIN-45 in
Caenorhabditis elegans [31][32][31,32]. A-Raf is the smallest subtype with a molecular weight of 68 kDa, C-Raf having a weight of 73 kDa, while B-Raf ranges from 75 to 100 kDa, this being attributable to variable splicing
[33][34][33,34].
The Human Protein Atlas expression database shows that C-Raf mRNA and A-Raf mRNA are predominantly present in skeletal muscle, bone marrow and the proximal digestive tract, while B-Raf mRNA is highly expressed in the retina, bone marrow and brain. Genetic and biochemical studies in
C. elegans and
D. melanogaster have demonstrated that Raf functions downstream of Ras and participates in cell proliferation, differentiation and development
[31][32][31,32].
The mammalian mitogen-activated protein kinase (MAPK) was first identified in mammalian cells and then in yeast
[35][36][35,36]. When adipocytes were briefly treated with insulin, a soluble serine/threonine kinase was rapidly activated, leading to increased phosphotyrosine content on microtubule-associated protein-2. Hence, it was initially named microtubule-associated protein-2 protein kinase (MAP-2 kinase). As it promotes cell cycle progression in response to insulin, growth factors and transforming proteins of oncogenic viruses, and phosphorylates a variety of protein substrates, it was renamed mitogen-activated protein kinase (MAPK) or extracellular signal-regulated kinase (ERK)
[35][37][38][35,37,38]. Two isoforms, ERK1 and ERK2, are encoded by the
MAPK3 and
MAPK1 genes, respectively. Activation of ERK1/2 requires phosphorylation at threonine and tyrosine residues and inactivation through dephosphorylation
[13][37][39][40][13,37,39,40]. Therefore, it is believed that a specific upstream kinase is responsible for phosphorylation and activation of ERK1/2
[41]. Subsequently, this kinase was found to be a dual-specificity kinase that phosphorylates threonine and tyrosine on ERK and is activated by nerve growth factor or epidermal growth factor, named MAP kinase kinase (MKK) or MAPK/ERK kinase (MEK)
[42][43][42,43]. The kinase were cloned by independent groups
[44][45][44,45]. Like ERK, MEK is also under negative regulation by dephosphorylation
[13]. Further studies of the kinase cascade have revealed that Raf is an upstream kinase that controls MEK activity by phosphorylation. Thus, Raf kinase was positioned as the first serine/threonine upstream kinase of the canonical MEK-ERK pathway in 1992
[3] and a direct effector of Ras in 1993
[4][46][47][4,46,47].
3. Role of Raf in Biology
Genetic knockouts of different Raf isoforms in mice all lead to embryonic lethality or severe growth retardation and abnormal development
[48][49][50][143,144,145]. Thus, A-Raf knockout mice were born alive but showed severe intestinal distension and neurological defects and died around postnatal day 20
[48][143]. B-Raf knockout mice succumbed in utero at embryonic day 12.5 due to massive bleeding in the body cavity, with severe vascular and neuronal abnormalities
[49][144]. The phenotypes are attributed to increased apoptosis of endothelial cells and endothelial precursor cells in embryos and large blood vessels
[51][146]. Ablation of C-Raf is fatal from embryonic day 10.5 to day 12.5, with dysplasia of the placenta, liver, hematopoietic organs, profound deafness and increased apoptosis of tissue cells
[50][52][53][54][145,147,148,149]. It has been shown that C-Raf plays a critical role in the regulation of cell proliferation and suppression of apoptosis during embryogenesis
[54][149]. A-Raf acts as a B-Raf effector and participates in Ras signaling when C-Raf is exhausted
[55][150]. Moreover, A-Raf stabilizes B-Raf–C-Raf interaction to maintain signaling efficiency, especially in the presence of Raf inhibitors
[55][150]. However, the cellular function of individual Raf is still less clear. Complex connections and interactions exist among them, which may depend on cell types or developmental stages.
Raf kinase family kinases play important roles in oncogenesis, inasmuch as they act as key effectors downstream of Ras, whose mutations account for oncogenesis in approximately 30% of human cancers, and as mediator for other oncogenes
[11][55][56][57][11,150,151,152]. B-Raf has attracted great interest since the report that it was found to be mutated in 66% of malignant melanomas in 2002
[58][153]. Over 100 mutations in B-Raf have been identified in cancer patients. Most B-Raf mutations are concentrated in two regions: the glycine-rich P loop of the N lobe, and the activation segment and flanking regions in the kinase domain
[59][154]. Among them, the most common mutation is a single amino acid substitution of valine 600 (V600, some reports designated V599) for glutamic acid, accounting for up to 90% of the mutations
[58][60][61][124,153,155]. While most of the mutations significantly increase the kinase activity, some (e.g., B-Raf G595R) exhibit impaired activity and cannot phosphorylate MEK directly
[62][156]. However, the B-Raf mutants with decreased kinase activity could hyper-stimulate the ERK pathway
[59][63][154,157]. Therefore, the oncogenic mechanism of B-Raf is fundamentally different from that of the constitutively activated v-Raf found in murine retrovirus
[64][158].
Although mutations of Raf-1 are much rarer in cancer than B-Raf, several studies have reported germline mutations of C-Raf in human diseases. For example, two mutations, S427G and I448V, are found in the kinase domain of C-Raf in patients with therapy-related acute myeloid leukemia
[65][159]. The mutation of S427G causes increased activity of the Raf/MEK/ERK pathway, while I448V mutation does not affect the kinase activity. This
res
earchtudy suggests that these germline mutations of C-Raf are predisposing factors for human neoplasia. In addition, mutation of C-Raf has been documented in “RASopathies”—a diverse collection of disorders caused by germline mutations in genes that code for the components or regulators of the RAS-RAF-MEK-ERK pathway. The disorders are characterized by postnatal short stature and neurocognitive delay, including neurofibromatosis type 1, Noonan syndrome, Noonan syndrome with multiple lentigines, cardio-facio-cutaneous syndrome and Legius syndrome
[66][67][68][160,161,162].
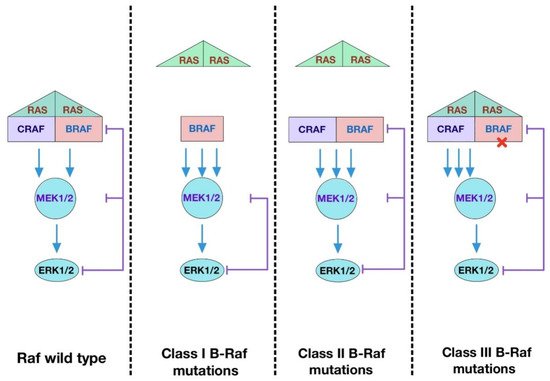
In terms of dimerization state, there are three major types of B-Raf mutations described in human cancers (
Figure 13). The most common one is the Class I with V600E mutation, which renders the kinase active as a monomer. The mutation mimics phosphorylation of the activation loop, leading to disruption of inactive conformation
[58][153]. Class II mutations, including K601E, L597Q, and G469A, cause spontaneous dimerization, resulting in the activation of the kinase. These mutations destabilize auto-inhibition by disrupting the inhibitory interaction of the activation loop with the Gly-rich loop and disable feedback suppression of Raf dimers
[69][163]. Class III mutations impair kinase activity toward MEK and adopt a tumor-specific mechanism by which the mutants transactivate endogenous C-Raf through phosphorylation of the activation loop by forming B-Raf–C-Raf heterodimers
[70][164]. Unlike Class I and Class II mutants, Class III mutants bind to Ras more tightly than wild-type B-Raf. Intriguingly, mutations equivalent to B-Raf V600E in C-Raf and A-Raf fail to produce oncogenic effects unless a negative charge is introduced into the NtA-region. These results support the notion that the regulation of C-Raf and A-Raf kinase domains is tighter because mutations at the two sites are required to confer transforming ability
[69][71][163,165].
Figure 13. Functional classes of B-Raf mutations. Class I B-Raf mutants contain V600E/D mutations in the activation loop which can signal as active monomers, independent of Ras. Class II B-Raf mutants are Ras-independent and signal as dimers. Class III B-Raf mutants have reduced kinase activity and drive the activation of ERK signaling by transactivating wild-type Raf which signals as mutant B-Raf–wild-type C-Raf dimers. These mutants require active Ras to trigger a signaling cascade.