 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | NORIKO FUNATO | -- | 2046 | 2022-05-19 04:26:20 | | | |
| 2 | Beatrix Zheng | Meta information modification | 2046 | 2022-05-19 09:54:00 | | | | |
| 3 | Beatrix Zheng | + 2 word(s) | 2048 | 2022-05-20 03:04:05 | | | | |
| 4 | Beatrix Zheng | + 264 word(s) | 2312 | 2022-05-27 05:53:15 | | |
Video Upload Options
TBX1, located on chromosome 22q11.21, encodes a T-box transcription factor and is a candidate gene for DiGeorge syndrome (DGS) and velocardiofacial syndrome (VCFS). Studies of Tbx1-mutant mice have provided insights into the underlying pathogenesis of DGS/VCFS and the knowledge to diagnose patients with DGS/VCFS. Genes, miRNAs, and epigenetics could change Tbx1 expression. Polymorphisms, variations, and mutations in TBX1 may induce the penetrance and severity of DGS/VCFS-like craniofacial phenotypes. The molecular basis of the variant sequence of TBX1 will further define how TBX1 contributes to the craniofacial and other phenotypes of DGS/VCFS. Since interactions with TBX1 and other molecules in transcriptional complexes or chromatin remodeling are crucial for TBX1 function, identifying and understanding these genetic and epigenetic modifiers individually for each patient may direct therapeutics to minimize the severity.
1. Introduction
2. Craniofacial Phenotypes of Patients with DGS/VCFS
| Phenotypes | Features | Frequency |
|---|---|---|
| Palatal anomalies | Overt cleft palate | 7–11% |
| Submucous cleft palate | 5–23% | |
| Bifid uvula | 5–10% | |
| Velopharyngeal insufficiency | 27–92% | |
| Dental anomalies | Tooth agenesis | 15% |
| Hypoplasia of primary teeth | 32% | |
| Hypoplasia of permanent teeth | 10% | |
| Enamel hypomineralization of primary teeth | 39% | |
| Enamel hypomineralization of permanent teeth | 41% | |
| Ear-nose-throat abnormalities | Hearing loss | 33–39% |
| Otitis media with effusion | 2% | |
| Tracheomalacia/laryngomalacia | 2% | |
| Laryngeal web | 1% | |
| Ocular abnormalities | Hooding of the upper lid | 41% |
| Ptosis | 9% | |
| Hooding of the lower lid | 6% | |
| Epicanthal folds | 3% | |
| Distichiasis | 3% | |
| Cranial base anomalies | Platybasia | 50–91% |
| Basilar impression | 3% | |
| Cervical spine anomalies | Atlas (C1) anomalies | 75% |
| Axis (C2) anomalies | 59% | |
| Fusion of C2–C3 | 34% |
| DGS/VCFS | Tbx1-Null Mice | |
|---|---|---|
| Cranium | Dolichocephaly | Small cranium |
| Abnormal skull morphology | Hypoplastic parietal bone | |
| Malar flattening | Hypoplastic interparietal bone | |
| Long face | Unfused cranial sutures between frontal and parietal bones | |
| Temporal bone hypoplasia | ||
| Absent zygomatic arch | ||
| Abnormal zygomatic arch morphology | ||
| Cranial Base | Platybasia | Abnormal fusion of the basioccipital and basisphenoid bones |
| Basilar impression | Abnormal presphenoid bone morphology | |
| Abnormal basioccipital bone morphology | ||
| Palate | Cleft palate | Cleft palate |
| Submucous cleft palate | Submucous cleft palate | |
| Bifid uvula | Bifid uvula | |
| Highly arched palate | ||
| Velopharyngeal insufficiency | ||
| Mandible | Retrognathia | Absent mandibular coronoid process |
| Short mandible | Short mandible | |
| Micrognathia | Micrognathia | |
| Teeth | Enamel hypoplasia | Abnormal upper incisor morphology |
| Single central incisor | Absent upper incisors | |
| Small teeth | ||
| Abnormality of the dentition | ||
| Carious teeth | ||
| Muscles | Pharyngeal hypotonia | Absent masseter muscle |
| Absent pterygoid muscle | ||
| Absent temporalis muscle | ||
| Eyes | Hypertelorism/telecanthus | Hypertelorism |
| Downslanted palpebral fissures | ||
| Proptosis | ||
| Strabismus | ||
| Abnormal eyelid morphology | ||
| Epicanthus | ||
| Microphthalmia | ||
| External Ears | Small earlobe | Ear lobe hypoplasia |
| Low-set ears | Lowered ear position | |
| Abnormally folded pinna | Abnormal ear shape | |
| Preauricular pit | Absent outer ear | |
| Anotia | ||
| Middle and Inner Ears | Chronic otitis media | Abnormal middle ear ossicle morphology |
| Conductive hearing loss | Absent middle ear ossicles | |
| Sensorineural hearing loss | Abnormal stapes morphology | |
| Auditory canal stenosis | Abnormal incus morphology | |
| Pulsatile tympanic membrane | Abnormal malleus morphology | |
| Thickened tympanic membrane | Absent stapes | |
| Tympanic membrane retraction | Abnormal external auditory canal morphology | |
| Decreased tympanic ring size | ||
| Nose | Prominent nasal bridge | Short snout |
| Abnormal nasal morphology | ||
| Underdeveloped nasal alae | ||
| Choanal atresia | ||
| Throat | Abnormal thorax morphology | Small thyroid cartilage |
| Abnormality of the pharynx | Small cricoid cartilage | |
| Abnormal thyroid cartilage morphology | ||
| Pharynx hypoplasia | ||
| Hyoid bones | Delayed development of the hyoid bone | Hyoid bone hypoplasia |
| Invisible hyoid ossification center | Abnormal hyoid bone morphology | |
| Cervical spine | Dysmorphic C1 | Abnormal cervical atlas (C1) morphology |
| Anterior arch cleft of C1 | Absent arcus anterior of C1 | |
| Open posterior arch C1 | ||
| Fusion of C1–C2 | ||
| Fusion of C2–C3 | ||
| Upswept C2 lamina | ||
| Platyspondyly | ||
| Others | Short clavicle | |
| References | [14][15][16][17][18][19][20][21][22][23][24][25][26] | [6][7][8][9][10][27][28][29][30][31][32][33][34] |
3. Genetics of DGS/VCFS
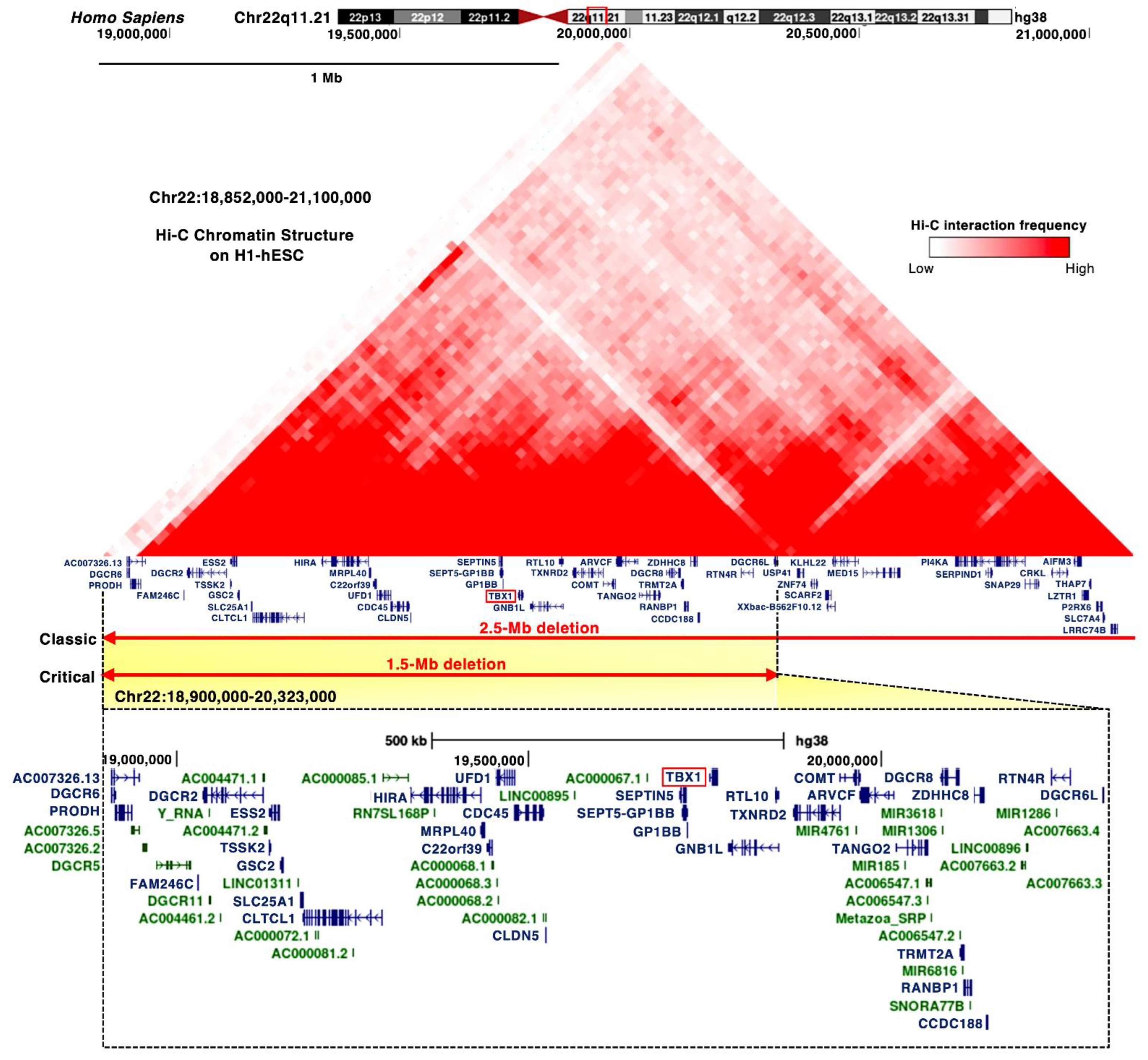
3.1. TBX1 Gene
| Mutation | Domain | Condition | Craniofacial Anomalies | References |
|---|---|---|---|---|
| c.89_284del | N-terminal | DiGeorge syndrome | Yes | ClinVar Variant: 971780 |
| c.199_224del | N-terminal | DiGeorge syndrome | Yes | ClinVar Variant: 949172 |
| c.292A>T | N-terminal | DiGeorge syndrome | Yes | ClinVar Variant: 526036 |
| c.385G>A | T-box | Tetralogy of Fallot | No | ClinVar Variant: 488618 |
| c.443T>A (F148Y) | T-box | Conotruncal anomaly face syndrome | Yes | [5] |
| c.503T>C | T-box | DiGeorge syndrome Velocardiofacial syndrome (Shprintzen syndrome) Tetralogy of Fallot |
Yes | ClinVar Variant: 973222 |
| c.569C > A (P190Q) | T-box | Congenital heart defects | No | [36] |
| c.582C>G (H194Q) | T-box | Velocardiofacial syndrome | Yes | [37] |
| c.928G>A (G310S) | C-terminal | DiGeorge syndrome | Yes | [5] |
| c.967_977dup AACCCCGTGGC | C-terminal | Thymic hypoplasia Postaxial polydactyly of the right fifth toe |
No | [38] |
| c.1158_1159delinsT | C-terminal | Hypoparathyroidism and hypocalcemia Facial asymmetry Deafness |
Yes | [39] |
| c.1223delC | C-terminal | Conotruncal anomaly face syndrome Velocardiofacial syndrome |
Yes | [5] |
| c.1253delA | C-terminal | DiGeorge syndrome | Yes | [40] |
| c.1320-1342del23bp | C-terminal | Velocardiofacial syndrome | Yes/No | [41] |
| c.1399-1428dup30 | C-terminal | Tetralogy of Fallot Scoliosis Facial asymmetry Upslanting palpebral fissures Absent pulmonary valve Isolated left pulmonary artery |
Yes | [42] |
3.2. DiGeorge Syndrome Critical Region (DGCR)
3.3. MicroRNAs
4. Craniofacial Phenotypes of DGS/VCFS Mouse Models
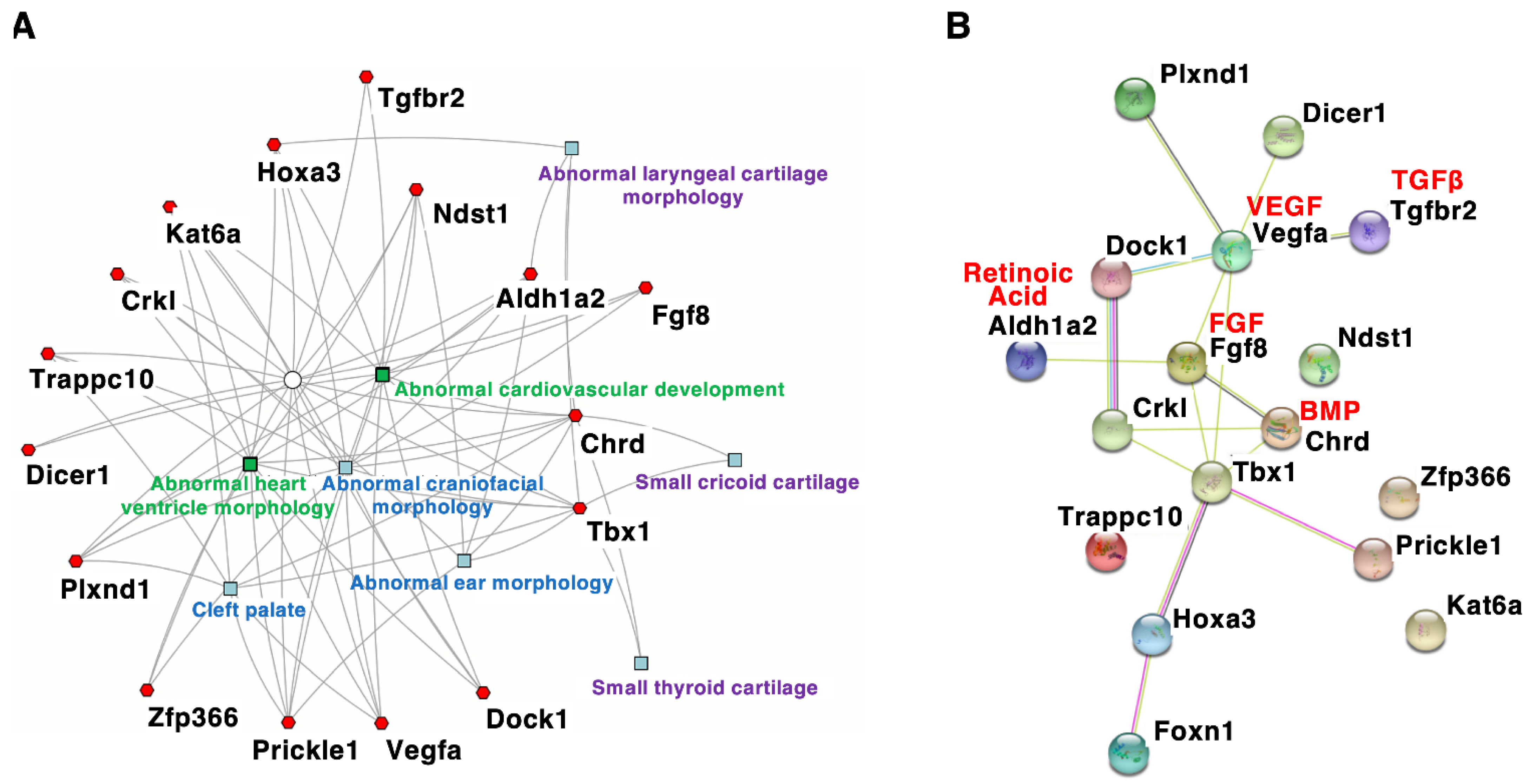
| Gene Symbol | Induced Mutation Type | Cranium | Palate | Teeth | Muscles | Ear-Nose-Throat | Hyoid Bones | Cardio-Vascular |
|---|---|---|---|---|---|---|---|---|
| Tbx1 | Null | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Chrd | Null | Yes | Yes | nr | nr | Yes | Yes | Yes |
| Tgfbr2 | Deletion (Wnt1-Cre) | Yes | Yes | nr | nr | nr | nr | Yes |
| Vegfa | Null | Yes | Yes | Yes | nr | nr | nr | Yes |
| Fgf8 | Hypomorphic allele | Yes | Yes | Yes | nr | Yes | Yes | Yes |
| Crkl | Null | Yes | nr | nr | nr | Yes | nr | Yes |
| Aldh1a2 | Hypomorphic allele | nr | nr | nr | nr | Yes | Yes | Yes |
| Hoxa3 | Null | nr | Yes | nr | Yes | Yes | Yes | Yes |
| Kat6a | Null | nr | Yes | nr | nr | Yes | nr | Yes |
| Dicer1 | Deletion (Wnt1-Cre) | Yes | nr | nr | nr | nr | nr | Yes |
| Plxnd1 | Single point mutation | nr | Yes | nr | nr | Yes | nr | Yes |
| Dock1 | Undefined | nr | nr | nr | nr | Yes | nr | Yes |
| Ndst1 | Single point mutation | nr | nr | nr | nr | Yes | nr | Yes |
| Prickle1 | Single point mutation | Yes | Yes | nr | nr | Yes | nr | Yes |
| Trappc10 | Undefined | Yes | Yes | nr | nr | nr | nr | Yes |
| Zfp366 | Single point mutation | nr | nr | nr | nr | Yes | nr | Yes |
| Foxn1 | Intragenic deletion | nr | nr | nr | nr | Yes | nr | Yes |
References
- Kaimal, V.; Bardes, E.E.; Tabar, S.C.; Jegga, A.G.; Aronow, B.J. ToppCluster: A multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 2010, 38, W96-102.
- Jerome, L.A.; Papaioannou, V.E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001, 27, 286–291.
- Lindsay, E.A.; Vitelli, F.; Su, H.; Morishima, M.; Huynh, T.; Pramparo, T.; Jurecic, V.; Ogunrinu, G.; Sutherland, H.F.; Scambler, P.J.; et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001, 410, 97–101.
- Merscher, S.; Funke, B.; Epstein, J.A.; Heyer, J.; Puech, A.; Lu, M.M.; Xavier, R.J.; Demay, M.B.; Russell, R.G.; Factor, S.; et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001, 104, 619–629.
- Hu, T.; Yamagishi, H.; Maeda, J.; McAnally, J.; Yamagishi, C.; Srivastava, D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development 2004, 131, 5491–5502.
- Liao, J.; Kochilas, L.; Nowotschin, S.; Arnold, J.S.; Aggarwal, V.S.; Epstein, J.A.; Brown, M.C.; Adams, J.; Morrow, B.E. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum. Mol. Genet. 2004, 13, 1577–1585.
- Zhang, Z.; Huynh, T.; Baldini, A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development 2006, 133, 3587–3595.
- Arnold, J.S.; Werling, U.; Braunstein, E.M.; Liao, J.; Nowotschin, S.; Edelmann, W.; Hebert, J.M.; Morrow, B.E. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development 2006, 133, 977–987.
- Arnold, J.S.; Braunstein, E.M.; Ohyama, T.; Groves, A.K.; Adams, J.C.; Brown, M.C.; Morrow, B.E. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum. Mol. Genet. 2006, 15, 1629–1639.
- Choi, M.; Klingensmith, J. Chordin is a modifier of tbx1 for the craniofacial malformations of 22q11 deletion syndrome phenotypes in mouse. PLoS Genet. 2009, 5, e1000395.
- Moraes, F.; Novoa, A.; Jerome-Majewska, L.A.; Papaioannou, V.E.; Mallo, M. Tbx1 is required for proper neural crest migration and to stabilize spatial patterns during middle and inner ear development. Mech. Dev. 2005, 122, 199–212.
- Funato, N.; Nakamura, M.; Richardson, J.A.; Srivastava, D.; Yanagisawa, H. Tbx1 regulates oral epithelial adhesion and palatal development. Hum. Mol. Genet. 2012, 21, 2524–2537.
- Funato, N.; Nakamura, M.; Richardson, J.A.; Srivastava, D.; Yanagisawa, H. Loss of Tbx1 induces bone phenotypes similar to cleidocranial dysplasia. Hum. Mol. Genet. 2015, 24, 424–435.
- Funato, N.; Srivastava, D.; Shibata, S.; Yanagisawa, H. TBX1 Regulates Chondrocyte Maturation in the Spheno-occipital Synchondrosis. J. Dent. Res. 2020, 99, 1182–1191.
- Bachiller, D.; Klingensmith, J.; Shneyder, N.; Tran, U.; Anderson, R.; Rossant, J.; De Robertis, E.M. The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development 2003, 130, 3567–3578.
- Wurdak, H.; Ittner, L.M.; Lang, K.S.; Leveen, P.; Suter, U.; Fischer, J.A.; Karlsson, S.; Born, W.; Sommer, L. Inactivation of TGFβ signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005, 19, 530–535.
- Stalmans, I.; Lambrechts, D.; De Smet, F.; Jansen, S.; Wang, J.; Maity, S.; Kneer, P.; von der Ohe, M.; Swillen, A.; Maes, C.; et al. VEGF: A modifier of the del22q11 (DiGeorge) syndrome? Nat. Med. 2003, 9, 173–182.
- Abu-Issa, R.; Smyth, G.; Smoak, I.; Yamamura, K.; Meyers, E.N. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development 2002, 129, 4613–4625.
- Guris, D.L.; Fantes, J.; Tara, D.; Druker, B.J.; Imamoto, A. Mice lacking the homologue of the human 22q11.2 gene CRLK phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet. 2001, 27, 293–298.
- Vermot, J.; Niederreither, K.; Garnier, J.M.; Chambon, P.; Dollé, P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1763–1768.
- Chisaka, O.; Capecchi, M.R. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature 1991, 350, 473–479.
- Voss, A.K.; Vanyai, H.K.; Collin, C.; Dixon, M.P.; McLennan, T.J.; Sheikh, B.N.; Scambler, P.; Thomas, T. MOZ Regulates the Tbx1 Locus, and Moz Mutation Partially Phenocopies DiGeorge Syndrome. Dev. Cell 2012, 23, 652–663.
- Sheehy, N.T.; Cordes, K.R.; White, M.P.; Ivey, K.N.; Srivastava, D. The neural crest-enriched microRNA miR-452 regulates epithelial-mesenchymal signaling in the first pharyngeal arch. Development 2010, 137, 4307–4316.
- Gershwin, M.E. DiGeorge syndrome: Congenital thymic hypoplasia. Animal model: Congenitally athymic (nude) mouse. Am. J. Pathol. 1977, 89, 809–812.
- Kobrynski, L.J.; Sullivan, K.E. Velocardiofacial syndrome, DiGeorge syndrome: The chromosome 22q11.2 deletion syndromes. Lancet 2007, 370, 1443–1452.
- Oberoi, S.; Vargervik, K. Velocardiofacial syndrome with single central incisor. Am. J. Med. Genet. A 2005, 132A, 194–197.
- Zhang, Z.; Huynh, T.; Baldini, A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development 2006, 133, 3587–3595.
- Arnold, J.S.; Werling, U.; Braunstein, E.M.; Liao, J.; Nowotschin, S.; Edelmann, W.; Hebert, J.M.; Morrow, B.E. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development 2006, 133, 977–987.
- Arnold, J.S.; Braunstein, E.M.; Ohyama, T.; Groves, A.K.; Adams, J.C.; Brown, M.C.; Morrow, B.E. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum. Mol. Genet. 2006, 15, 1629–1639.
- Choi, M.; Klingensmith, J. Chordin is a modifier of tbx1 for the craniofacial malformations of 22q11 deletion syndrome phenotypes in mouse. PLoS Genet. 2009, 5, e1000395.
- Moraes, F.; Novoa, A.; Jerome-Majewska, L.A.; Papaioannou, V.E.; Mallo, M. Tbx1 is required for proper neural crest migration and to stabilize spatial patterns during middle and inner ear development. Mech. Dev. 2005, 122, 199–212.
- Funato, N.; Nakamura, M.; Richardson, J.A.; Srivastava, D.; Yanagisawa, H. Tbx1 regulates oral epithelial adhesion and palatal development. Hum. Mol. Genet. 2012, 21, 2524–2537.
- Funato, N.; Nakamura, M.; Richardson, J.A.; Srivastava, D.; Yanagisawa, H. Loss of Tbx1 induces bone phenotypes similar to cleidocranial dysplasia. Hum. Mol. Genet. 2015, 24, 424–435.
- Funato, N.; Srivastava, D.; Shibata, S.; Yanagisawa, H. TBX1 Regulates Chondrocyte Maturation in the Spheno-occipital Synchondrosis. J. Dent. Res. 2020, 99, 1182–1191.
- Solot, C.B.; Sell, D.; Mayne, A.; Baylis, A.L.; Persson, C.; Jackson, O.; McDonald-McGinn, D.M. Speech-Language Disorders in 22q11.2 Deletion Syndrome: Best Practices for Diagnosis and Management. Am. J. Speech-Lang. Pathol. 2019, 28, 984–999.
- Jaouadi, A.; Tabebi, M.; Abdelhedi, F.; Abid, D.; Kamoun, F.; Chabchoub, I.; Maatoug, S.; Doukali, H.; Belghuith, N.; Ksentini, M.A.; et al. A novel TBX1 missense mutation in patients with syndromic congenital heart defects. Biochem. Biophys. Res. Commun. 2018, 499, 563–569.
- Zweier, C.; Sticht, H.; Aydin-Yaylagül, I.; Campbell, C.E.; Rauch, A. Human TBX1 Missense Mutations Cause Gain of Function Resulting in the Same Phenotype as 22q11.2 Deletions. Am. J. Hum. Genet. 2007, 80, 510–517.
- Hasegawa, K.; Tanaka, H.; Higuchi, Y.; Hayashi, Y.; Kobayashi, K.; Tsukahara, H. Novel heterozygous mutation in TBX1 in an infant with hypocalcemic seizures. Clin. Pediatr. Endocrinol. 2018, 27, 159–164.
- Alghamdi, M.; Al Khalifah, R.; Al Homyani, D.K.; Alkhamis, W.H.; Arold, S.T.; Ekhzaimy, A.; El-Wetidy, M.; Kashour, T.; Halwani, R. A novel TBX1 variant causing hypoparathyroidism and deafness. J. Endocr. Soc. 2020, 4, bvz028.
- Ogata, T.; Niihori, T.; Tanaka, N.; Kawai, M.; Nagashima, T.; Funayama, R.; Nakayama, K.; Nakashima, S.; Kato, F.; Fukami, M.; et al. TBX1 mutation identified by exome sequencing in a Japanese family with 22q11.2 deletion syndrome-like craniofacial features and hypocalcemia. PLoS ONE 2014, 9, e91598.
- Paylor, R.; Glaser, B.; Mupo, A.; Ataliotis, P.; Spencer, C.; Sobotka, A.; Sparks, C.; Choi, C.H.; Oghalai, J.; Curran, S.; et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 7729–7734.
- Rauch, R.; Hofbeck, M.; Zweier, C.; Koch, A.; Zink, S.; Trautmann, U.; Hoyer, J.; Kaulitz, R.; Singer, H.; Rauch, A. Comprehensive genotype-phenotype analysis in 230 patients with tetralogy of Fallot. J. Med. Genet. 2010, 47, 321–331.
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.-S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760.
- Edelmann, L.; Stankiewicz, P.; Spiteri, E.; Pandita, R.K.; Shaffer, L.; Lupski, J.R.; Morrow, B.E.; Lupski, J. Two functional copies of the DGCR6 gene are present on human chromosome 22q11 due to a duplication of an ancestral locus. Genome Res. 2001, 11, 208–217.
- Hierck, B.P.; Molin, D.G.M.; Boot, M.J.; Poelmann, R.E.; Gittenberger-De Groot, A.C. A chicken model for DGCR6 as a modifier gene in the DiGeorge critical region. Pediatr. Res. 2004, 56, 440–448.
- Gao, S.; Moreno, M.; Eliason, S.; Cao, H.; Li, X.; Yu, W.; Bidlack, F.B.; Margolis, H.C.; Baldini, A.; Amendt, B.A. TBX1 protein interactions and microRNA-96-5p regulation controls cell proliferation during craniofacial and dental development: Implications for 22q11.2 deletion syndrome. Hum. Mol. Genet. 2015, 24, 2330–2348.
- Sun, H.; Jiang, P. MicroRNA-451a acts as tumor suppressor in cutaneous basal cell carcinoma. Mol. Genet. Genom. Med. 2018, 6, 1001–1009.
- Wang, J.; Bai, Y.; Li, H.; Greene, S.B.; Klysik, E.; Yu, W.; Schwartz, R.J.; Williams, T.J.; Martin, J.F. MicroRNA-17-92, a direct Ap-2alpha transcriptional target, modulates T-box factor activity in orofacial clefting. PLoS Genet. 2013, 9, e1003785.

