TBX1, located on chromosome 22q11.21, encodes a T-box transcription factor and is a candidate gene for DiGeorge syndrome (DGS) and velocardiofacial syndrome (VCFS). Studies of Tbx1-mutant mice have provided insights into the underlying pathogenesis of DGS/VCFS and the knowledge to diagnose patients with DGS/VCFS. Genes, miRNAs, and epigenetics could change Tbx1 expression. Polymorphisms, variations, and mutations in TBX1 may induce the penetrance and severity of DGS/VCFS-like craniofacial phenotypes. The molecular basis of the variant sequence of TBX1 will further define how TBX1 contributes to the craniofacial and other phenotypes of DGS/VCFS. Since interactions with TBX1 and other molecules in transcriptional complexes or chromatin remodeling are crucial for TBX1 function, identifying and understanding these genetic and epigenetic modifiers individually for each patient may direct therapeutics to minimize the severity.
1. Introduction
The 22q11.2 deletion syndrome is one of the most common chromosomal microdeletions, affecting approximately 1 in 4000 live births in humans [
1]. A 1.5 to 2.5 Mb hemizygous deletion of chromosome 22q11.2 causes DiGeorge syndrome (DGS; OMIM #188400) and velocardiofacial syndrome (VCFS or Shprintzen VCF syndrome; OMIM #192430) [
2]. DGS/VCFS appears to be a genomic disorder distinct from 22q11.2 distal deletion syndrome (OMIM #611867). The clinical phenotype of DGS/VCFS is a complex and variable congenital disability, including cardiovascular defects, thymic hypoplasia, parathyroid hypoplasia, and craniofacial malformations [
3]. Craniofacial malformations occur in approximately 60% of patients with DGS/VCFS [
4].
TBX1, located on chromosome 22q11.21, encodes a T-box transcription factor and is considered a candidate gene for DGS/VCFS since mutations in
TBX1 have been found in patients with DGS/VCFS [
5]. Heterozygous
Tbx1-mutant (
Tbx1+/−) mice exhibit DGS/VCFS-related cardiovascular, parathyroid, and thymic phenotypes, suggesting that
TBX1 dosage is critical for cardiovascular, parathyroid and thymic development [
6,
7,
8,
9].
Tbx1-null mice exhibit the most clinical features of DGS/VCFS, including craniofacial phenotypes, while
Tbx1+/− mice exhibit no significant craniofacial phenotypes [
6,
7,
8,
9,
10].
There have been some excellent reviews on genetics and cardiovascular anomalies of DGS/VCFS [
3,
11,
12,
13]. However, information on the craniofacial anomalies of DGS/VCFS is limited. This review focuses on these phenotypes and summarizes the current understanding of the genetic factors that impact DGS/VCFS-related phenotypes. We also review DGS/VCFS mouse models that have been designed to better understand the pathogenic processes of DGS/VCFS.
2. Craniofacial Phenotypes of Patients with DGS/VCFS
Patients with DGS/VCFS manifest craniofacial anomalies involving the cranium, cranial base, jaws, pharyngeal muscles, ear-nose-throat, palate, teeth, and cervical spine (
Table 1 and
Table 2). Frequently observed craniofacial phenotypes include velopharyngeal insufficiency (27–92%), enamel hypomineralization (39–41%), hearing loss (33–39%), platybasia (50–91%), and cervical spine anomalies (75%) (
Table 1). Delayed development of the hyoid bone has also been reported [
14,
15].
Table 2. Craniofacial and skeletal phenotypes of DGS/VCFS and Tbx1-null mice.
| |
DGS/VCFS |
Tbx1-Null Mice |
| Cranium |
Dolichocephaly |
Small cranium |
| |
Abnormal skull morphology |
Hypoplastic parietal bone |
| |
Malar flattening |
Hypoplastic interparietal bone |
| |
Long face |
Unfused cranial sutures between frontal and parietal bones |
| |
|
Temporal bone hypoplasia |
| |
|
Absent zygomatic arch |
| |
|
Abnormal zygomatic arch morphology |
| Cranial Base |
Platybasia |
Abnormal fusion of the basioccipital and basisphenoid bones |
| |
Basilar impression |
Abnormal presphenoid bone morphology |
| |
|
Abnormal basioccipital bone morphology |
| Palate |
Cleft palate |
Cleft palate |
| |
Submucous cleft palate |
Submucous cleft palate |
| |
Bifid uvula |
Bifid uvula |
| |
Highly arched palate |
|
| |
Velopharyngeal insufficiency |
|
| Mandible |
Retrognathia |
Absent mandibular coronoid process |
| |
Short mandible |
Short mandible |
| |
Micrognathia |
Micrognathia |
| Teeth |
Enamel hypoplasia |
Abnormal upper incisor morphology |
| |
Single central incisor |
Absent upper incisors |
| |
Small teeth |
|
| |
Abnormality of the dentition |
|
| |
Carious teeth |
|
| Muscles |
Pharyngeal hypotonia |
Absent masseter muscle |
| |
|
Absent pterygoid muscle |
| |
|
Absent temporalis muscle |
| Eyes |
Hypertelorism/telecanthus |
Hypertelorism |
| |
Downslanted palpebral fissures |
|
| |
Proptosis |
|
| |
Strabismus |
|
| |
Abnormal eyelid morphology |
|
| |
Epicanthus |
|
| |
Microphthalmia |
|
| External Ears |
Small earlobe |
Ear lobe hypoplasia |
| |
Low-set ears |
Lowered ear position |
| |
Abnormally folded pinna |
Abnormal ear shape |
| |
Preauricular pit |
Absent outer ear |
| |
|
Anotia |
| Middle and Inner Ears |
Chronic otitis media |
Abnormal middle ear ossicle morphology |
| |
Conductive hearing loss |
Absent middle ear ossicles |
| |
Sensorineural hearing loss |
Abnormal stapes morphology |
| |
Auditory canal stenosis |
Abnormal incus morphology |
| |
Pulsatile tympanic membrane |
Abnormal malleus morphology |
| |
Thickened tympanic membrane |
Absent stapes |
| |
Tympanic membrane retraction |
Abnormal external auditory canal morphology |
| |
|
Decreased tympanic ring size |
| Nose |
Prominent nasal bridge |
Short snout |
| |
Abnormal nasal morphology |
|
| |
Underdeveloped nasal alae |
|
| |
Choanal atresia |
|
| Throat |
Abnormal thorax morphology |
Small thyroid cartilage |
| |
Abnormality of the pharynx |
Small cricoid cartilage |
| |
|
Abnormal thyroid cartilage morphology |
| |
|
Pharynx hypoplasia |
| Hyoid bones |
Delayed development of the hyoid bone |
Hyoid bone hypoplasia |
| |
Invisible hyoid ossification center |
Abnormal hyoid bone morphology |
| Cervical spine |
Dysmorphic C1 |
Abnormal cervical atlas (C1) morphology |
| |
Anterior arch cleft of C1 |
Absent arcus anterior of C1 |
| |
Open posterior arch C1 |
|
| |
Fusion of C1–C2 |
|
| |
Fusion of C2–C3 |
|
| |
Upswept C2 lamina |
|
| |
Platyspondyly |
|
| Others |
|
Short clavicle |
| References |
[14,15,16,17,18,19,20,21,22,23,24,25,26,27,28] |
[6,7,8,9,10,29,30,31,32,33,34,35,36] |
In addition to morphological anomalies, infants and young children with DGS/VCFS often exhibit a high prevalence of functional difficulties in feeding and speech/language associated with cleft palate, laryngeal anomalies, and velopharyngeal dysfunction [
37]. Even after cleft palate closure, children with DGS/VCFS sometimes present communication disorders related to speech-language problems, such as articulation disorders of speech sounds and vocal disorders [
37]. They exhibit slower language acquisition than those with other disorders that may be associated with abnormal muscle development.
3. Genetics of DGS/VCFS
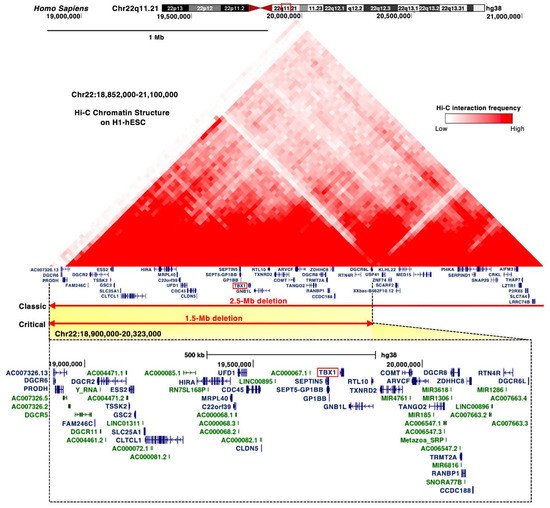
DGS/VCFS is caused by a 1.5 to 2.5 Mb hemizygous deletion of chromosome 22q11.2 (Figure 1). Chromosomal microdeletions at 10p14-p13 (the DGS2 locus) in patients with DGS/VCFS phenotypes are defined as the DGS/VCFS complex 2. In this review, we focus on the 22q11.2 locus, its associated genes, and miRNAs.
Figure 1. Proximal deletions of chromosome 22q11.2 are responsible for the clinical features of DGS/VCFS. Snapshot of the UCSC Genome Browser (
http://genome.ucsc.edu accessed on 3 August 2021) in the hg38 assembly showing the genomic context in the proximal deletions of chromosome 22q11.2. Top, the 25 kb resolution Hi-C data in H1 human embryonic stem cell line (H1-hESC)
[1]. Bottom, the coding (blue) and noncoding RNAs (green), including miRNAs and long noncoding RNAs, are shown.
Most of the chromosomal deletions of the 22q11.2 locus are de novo, but inherited deletions of the 22q11.2 locus have been reported in 6–28% of patients as autosomal dominant [
16,
17]. The majority of clinical phenotypes of DGS/VCFS are caused by proximal 1.5 Mb microdeletions [
3,
22], resulting in a hemizygosity of approximately 30 coding genes, including
DGCR6, PRODH, DGCR2, ESS2, TSSK2, GSC2, FAM246C, SLC25A1, CLTCL1, UFD1, HIRA, CDC45, MRPL40, C22orf39, CLDN5, TBX1, SEPTIN5, SEPT5-GP1BB, GP1BB, GNB1L, RTL10, TXNRD2, COMT, ARVCF, TANGO2, TRMT2A, RANBP1, CCDC188, DGCR8, ZDHHC8, RTN4R, DGCR6L, and
C007326, as well as microRNAs (miRNAs) and long noncoding RNAs (
Figure 1). The Hi-C chromatin structure of the 1.5 Mb region indicates interactions between these loci and their neighboring regions (
Figure 1).
3.1. TBX1 Gene
The proximal deletion of 1.5 Mb on the 22q11.2 locus includes
TBX1 (
Figure 1).
TBX1 is considered a candidate gene of DGS/VCFS because haploinsufficiency of
TBX1 leads to the typical phenotypes of DGS/VCFS, conotruncal anomaly face syndrome (OMIM #217095), and tetralogy of Fallot (OMIM #187500) (
Table 3). Identical mutations in
TBX1 present among patients resulted in distinct phenotypes, suggesting that genetic and epigenetic changes or environmental factors are involved in the clinical phenotypes [
5]. Further investigation is required to confirm that the variants cause DGS/VCFS and how they impact the phenotypes.
Table 3. DGS/VCFS-associated variants of TBX1.
| Mutation |
Domain |
Condition |
Craniofacial Anomalies |
References |
| c.89_284del |
N-terminal |
DiGeorge syndrome |
Yes |
ClinVar Variant: 971780 |
| c.199_224del |
N-terminal |
DiGeorge syndrome |
Yes |
ClinVar Variant: 949172 |
| c.292A>T |
N-terminal |
DiGeorge syndrome |
Yes |
ClinVar Variant: 526036 |
| c.385G>A |
T-box |
Tetralogy of Fallot |
No |
ClinVar Variant: 488618 |
| c.443T>A (F148Y) |
T-box |
Conotruncal anomaly face syndrome |
Yes |
[5] |
| c.503T>C |
T-box |
DiGeorge syndrome
Velocardiofacial syndrome
(Shprintzen syndrome)
Tetralogy of Fallot |
Yes |
ClinVar Variant: 973222 |
| c.569C > A (P190Q) |
T-box |
Congenital heart defects |
No |
[38] |
| c.582C>G (H194Q) |
T-box |
Velocardiofacial syndrome |
Yes |
[39] |
| c.928G>A (G310S) |
C-terminal |
DiGeorge syndrome |
Yes |
[5] |
| c.967_977dup AACCCCGTGGC |
C-terminal |
Thymic hypoplasia
Postaxial polydactyly of the right fifth toe |
No |
[40] |
| c.1158_1159delinsT |
C-terminal |
Hypoparathyroidism and hypocalcemia
Facial asymmetry
Deafness |
Yes |
[41] |
| c.1223delC |
C-terminal |
Conotruncal anomaly face syndrome
Velocardiofacial syndrome |
Yes |
[5] |
| c.1253delA |
C-terminal |
DiGeorge syndrome |
Yes |
[42] |
| c.1320-1342del23bp |
C-terminal |
Velocardiofacial syndrome |
Yes/No |
[43] |
| c.1399-1428dup30 |
C-terminal |
Tetralogy of Fallot
Scoliosis
Facial asymmetry
Upslanting palpebral fissures
Absent pulmonary valve
Isolated left pulmonary artery |
Yes |
[44] |
3.2. DiGeorge Syndrome Critical Region (DGCR)
DGCR8, DGCR6, and DGCR6L map to the commonly deleted 1.5 Mb region in DGS/VCFS (
Figure 1). DGCR8 is a nuclear miRNA-binding protein required for miRNA biogenesis. Dgcr8 haploinsufficiency in mice reduces the expression of miRNAs in the brain [
45]. DGCR6 and DGCR6L genes encode a protein with a sequence similar to the Drosophila gonadal [
46]. In a chicken model, targeting DGCR6 function resulted in a vascular phenotype [
47]. Attenuation of DGCR6 affects the expression of three genes localized within the 1.5 Mb region, upregulating the expression of TBX1 and UFD1 and reducing the expression of HIRA in the heart and pharyngeal arches of the chicken embryos [
47]. Thus, the haploinsufficiency of DGCR8 or DGCR6 may be linked to DGS/VCFS phenotypes when targeting DGS/VCFS-related genes and miRNAs.
3.3. MicroRNAs
The deleted 1.5 Mb on the 22q11.2 locus includes several miRNAs, such as miR-185, miR-4716, miR-3618, miR-1286, miR-1306, and miR-6816 (
Figure 1). The TargetScan miRNA target prediction program (
http://www.targetscan.org accessed on 3 August 2021) identified that the 3′ UTR of
TBX1 includes conserved sites for miR-183-5p, miR-96-5p, miR-1271-5p, miR-182-5p, miR-144-3p, miR-139-5p, miR-101-3p, and miR-451. Two miRNAs were confirmed to target the 3′ UTR of
TBX1. miR-96-5p represses
Tbx1 expression and, in turn, TBX1 suppresses the promoter activity and expression of miR-96 [
48]. miR-451a, a tumor suppressor, also directly targets
TBX1 [
49]. The expression of this gene is upregulated in cutaneous basal cell carcinoma, inversely to miR-451a [
49]. miR-17-92 fine-tunes the expression of
Tbx1 in craniofacial development, suggesting miR-17-92 as a candidate genetic modifier for
Tbx1 [
50]. Thus, miRNAs both inside and outside the 22q11.2 locus may influence the severity of the clinical phenotypes of DGS/VCFS.
4. Current Insights
The penetrance and severity of congenital anomalies are related to genetic and environmental factors. Recent studies have revealed the function of TBX1 and modifiers that impact the severity and penetrance of DGS/VCFS. Studies of DGS/VCFS mouse models have provided insights into signaling pathways and genes that interact with TBX1 and/or affect the DGS/VCFS phenotypes. In addition, mouse models with DGS/VCFS may help us to identify additional DGS/VCFS-related phenotypes. For example, there is potential to examine the phenotypes of cranial synchondroses, cranium, zygomatic arches, and pharyngeal muscles in DGS/VCFS patients. We also noted that information about ocular phenotypes in
Tbx1-mutant mice is limited, although these anomalies in patients with DGS/VCFS have been reported [
16,
17]. Crosstalk with key embryonic signals, especially bone morphogenetic protein (BMP), transforming growth factor (TGF)β, vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), Retinoic Acid, and Sonic Hedgehog (SHH), critically regulates DGS/VCFS-related pharyngeal development. Genes involved in these signaling pathways may modify the phenotypic spectrum of DGS/VCFS. Given the broad spectrum of DGS/VCFS disease phenotypes, other genes essential to craniofacial development could modify the phenotypic spectrum. Genetically engineered mice are useful for studying disease phenotypes; however, ablation of essential genes involved in cardiovascular development may cause early embryonic lethality, which would prevent observation of craniofacial phenotypes. For example, ablation of
Ufd1, whose human ortholog has been mapped to the 1.5 Mb region, causes early embryonic lethality before organogenesis in mice [
116]. It is also essential to identify novel proteins that interact with TBX1 and examine whether interacting partners may influence the phenotypes of mouse models.
This entry is adapted from the peer-reviewed paper 10.3390/jdb10020018