 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Teresa Zoladek | -- | 4114 | 2022-05-18 11:22:45 | | | |
| 2 | Beatrix Zheng | + 1 word(s) | 4115 | 2022-05-18 12:09:52 | | |
Video Upload Options
Mutations in human VPS13A-D genes result in rare neurological diseases, including chorea-acanthocytosis (ChAc). The pathogenesis of these diseases is poorly understood, and no effective treatment is available. As VPS13 genes are evolutionarily conserved, the effects of the pathogenic mutations could be studied in model organisms, including yeast, where one VPS13 gene is present. Here, the researchers summarize advancements obtained using yeast. In recent studies, vps13Δ and vps13-I2749 yeast mutants, which are models of chorea-acanthocytosis, were used to screen for multicopy and chemical suppressors. Two of the suppressors, a fragment of the MYO3 and RCN2 genes, act by downregulating calcineurin activity. In addition, vps13Δ suppression was achieved by using calcineurin inhibitors. The other group of multicopy suppressors were genes: FET4, encoding iron transporter, and CTR1, CTR3 and CCC2, encoding copper transporters. Mechanisms of their suppression rely on causing an increase in the intracellular iron content. Moreover, among the identified chemical suppressors were copper ionophores, which require a functional iron uptake system for activity, and flavonoids, which bind iron. These findings point at areas for further investigation in a higher eukaryotic model of VPS13-related diseases and to new therapeutic targets: calcium signalling and copper and iron homeostasis. Furthermore, the identified drugs are interesting candidates for drug repurposing for these diseases.
1. Introduction
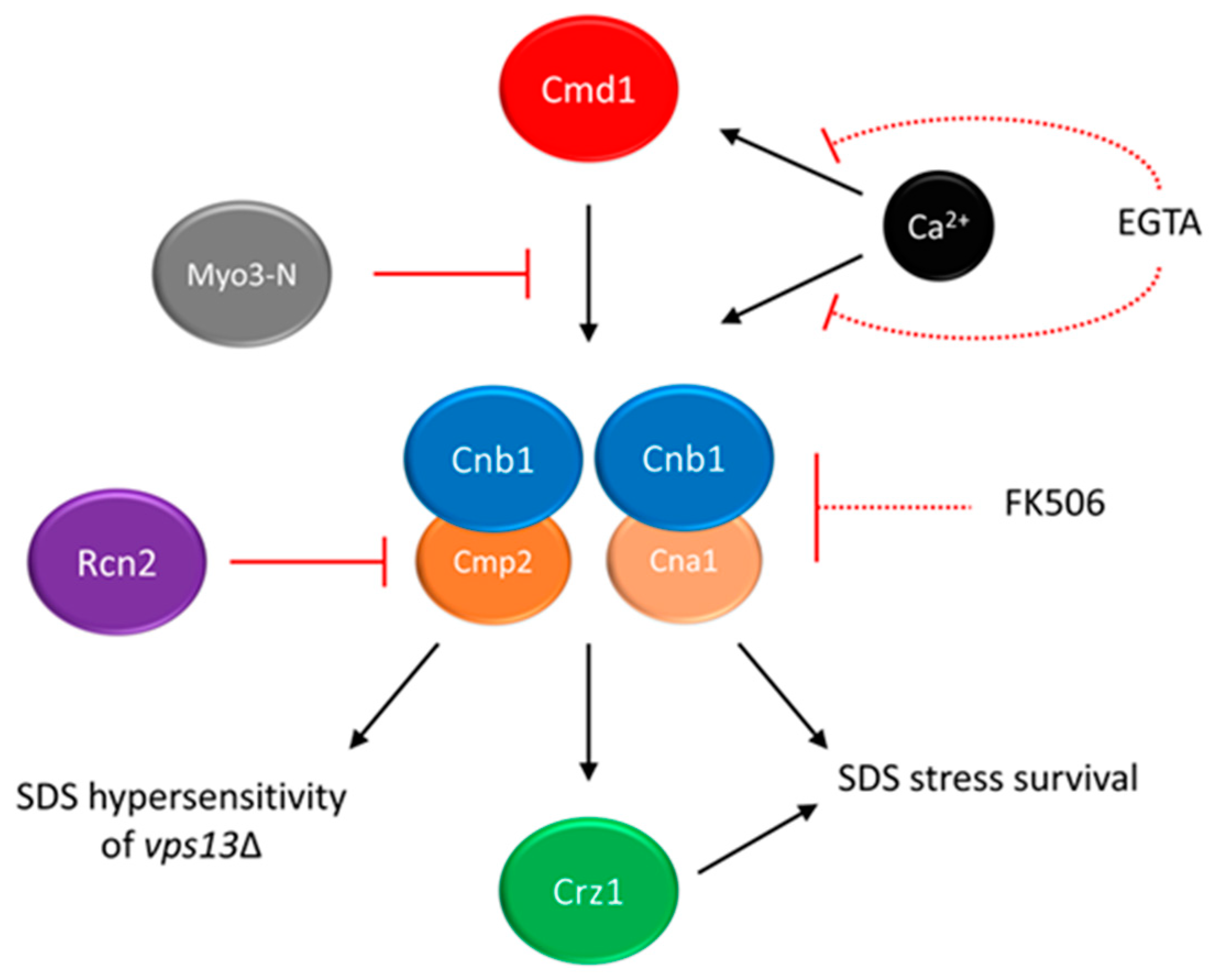
2. Calcium Signalling as a Potential Target for Drug Intervention in VPS13-Dependent Neurodegenerative Diseases
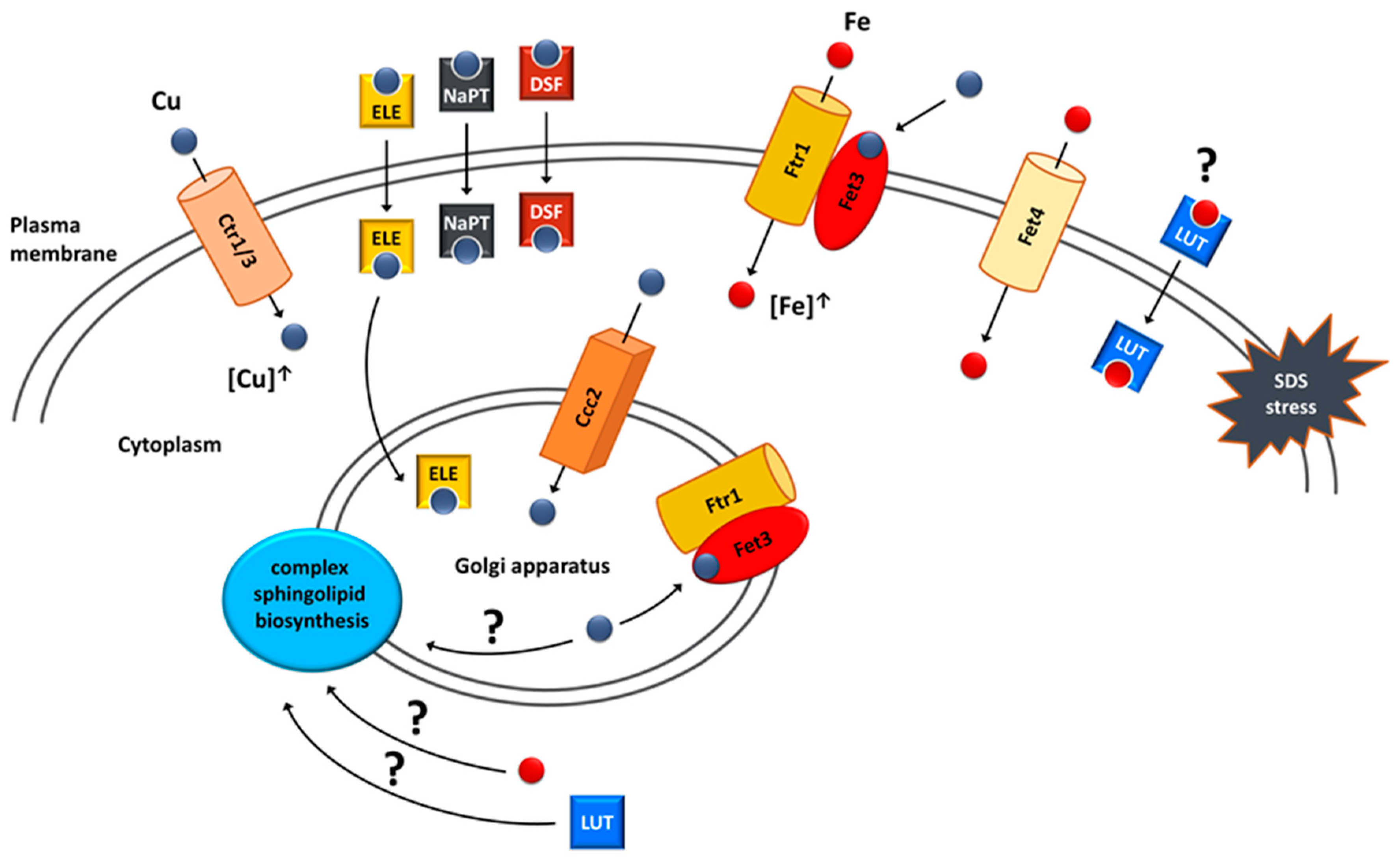
3. Copper and Iron Homeostasis as a Potential Target for Treatment of VPS13-Dependent Neurodegenerative Diseases
References
- Roessler, H.I.; Knoers, N.V.A.M.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharmacol. Sci. 2021, 42, 255–267.
- Bhattacharya, K.; Balasubramaniam, S.; Choy, Y.S.; Fietz, M.; Fu, A.; Jin, D.K.; Kim, O.H.; Kosuga, M.; Kwun, Y.H.; Inwood, A.; et al. Overcoming the barriers to diagnosis of Morquio A syndrome. Orphanet J. Rare Dis. 2014, 9, 192.
- Blöß, S.; Klemann, C.; Rother, A.K.; Mehmecke, S.; Schumacher, U.; Mücke, U.; Mücke, M.; Stieber, C.; Klawonn, F.; Kortum, X.; et al. Diagnostic needs for rare diseases and shared prediagnostic phenomena: Results of a German-wide expert Delphi survey. PLoS ONE 2017, 12, e0172532.
- Bouwman, M.G.; Teunissen, Q.G.; Wijburg, F.A.; Linthorst, G.E. ‘Doctor Google’ ending the diagnostic odyssey in lysosomal storage disorders: Parents using internet search engines as an efficient diagnostic strategy in rare diseases. Arch. Dis. Child. 2010, 95, 642–644.
- Hallal, C.; Kieling, C.O.; Nunes, D.L.; Ferreira, C.T.; Peterson, G.; Barros, S.G.; Arruda, C.A.; Fraga, J.C.; Goldani, H.A. Diagnosis, misdiagnosis, and associated diseases of achalasia in children and adolescents: A twelve-year single center experience. Pediatr. Surg. Int. 2012, 28, 1211–1217.
- Kishnani, P.S.; Amartino, H.M.; Lindberg, C.; Miller, T.M.; Wilson, A.; Keutzer, J.; Advisors, P.R.B.o. Timing of diagnosis of patients with Pompe disease: Data from the Pompe registry. Am. J. Med. Genet. A 2013, 161A, 2431–2443.
- Prencipe, N.; Floriani, I.; Guaraldi, F.; Di Giacomo, S.V.; Cannavo, S.; Arnaldi, G.; Berton, A.; Torri, V.; Spinello, M.; Arvat, E.; et al. ACROSCORE: A new and simple tool for the diagnosis of acromegaly, a rare and underdiagnosed disease. Clin. Endocrinol. 2016, 84, 380–385.
- Guffon, N.; Heron, B.; Chabrol, B.; Feillet, F.; Montauban, V.; Valayannopoulos, V. Diagnosis, quality of life, and treatment of patients with Hunter syndrome in the French healthcare system: A retrospective observational study. Orphanet J. Rare Dis. 2015, 10, 43.
- Pierucci, P.; Lenato, G.M.; Suppressa, P.; Lastella, P.; Triggiani, V.; Valerio, R.; Comelli, M.; Salvante, D.; Stella, A.; Resta, N.; et al. A long diagnostic delay in patients with Hereditary Haemorrhagic Telangiectasia: A questionnaire-based retrospective study. Orphanet J. Rare Dis. 2012, 7, 33.
- Wu, Y.; Xu, Y.Y.; Gao, Y.; Li, J.M.; Liu, X.W.; Wang, M.Q.; Deng, H.; Xiao, L.L.; Ren, H.B.; Xiong, B.T.; et al. Deep brain stimulation for chorea-acanthocytosis: A systematic review. Neurosurg. Rev. 2022; online ahead of print.
- Dobson-Stone, C.; Rampoldi, L.; Bader, B.; Walker, R.H.; Danek, A.; Monaco, A.P. Chorea-Acanthocytosis; University of Washington: Seattle, WA, USA, 2019.
- Stanslowsky, N.; Reinhardt, P.; Glass, H.; Kalmbach, N.; Naujock, M.; Hensel, N.; Lübben, V.; Pal, A.; Venneri, A.; Lupo, F.; et al. Neuronal Dysfunction in iPSC-Derived Medium Spiny Neurons from Chorea-Acanthocytosis Patients Is Reversed by Src Kinase Inhibition and F-Actin Stabilization. J. Neurosci. 2016, 36, 12027–12043.
- De Franceschi, L.; Tomelleri, C.; Matte, A.; Brunati, A.M.; Bovee-Geurts, P.H.; Bertoldi, M.; Lasonder, E.; Tibaldi, E.; Danek, A.; Walker, R.H.; et al. Erythrocyte membrane changes of chorea-acanthocytosis are the result of altered Lyn kinase activity. Blood 2011, 118, 5652–5663.
- Peikert, K.; Glaß, H.; Federti, E.; Matte, A.; Pelzl, L.; Akgün, K.; Ziemssen, T.; Ordemann, R.; Lang, F.; Patients, T.N.F.T.; et al. Targeting Lyn Kinase in Chorea-Acanthocytosis: A Translational Treatment Approach in a Rare Disease. J. Pers. Med. 2021, 11, 392.
- Federti, E.; Matte, A.; Riccardi, V.; Peikert, K.; Alper, S.L.; Danek, A.; Walker, R.H.; Siciliano, A.; Iatcenko, I.; Hermann, A.; et al. Adaptative Up-Regulation of PRX2 and PRX5 Expression Characterizes Brain from a Mouse Model of Chorea-Acanthocytosis. Antioxidants 2021, 11, 76.
- Peikert, K.; Federti, E.; Matte, A.; Constantin, G.; Pietronigro, E.C.; Fabene, P.F.; Defilippi, P.; Turco, E.; Del Gallo, F.; Pucci, P.; et al. Therapeutic targeting of Lyn kinase to treat chorea-acanthocytosis. Acta Neuropathol. Commun. 2021, 9, 81.
- Dziurdzik, S.K.; Bean, B.D.M.; Davey, M.; Conibear, E. A VPS13D spastic ataxia mutation disrupts the conserved adaptor-binding site in yeast Vps13. Hum. Mol. Genet. 2020, 29, 635–648.
- Park, J.S.; Hollingsworth, N.M.; Neiman, A.M. Genetic Dissection of Vps13 Regulation in Yeast Using Disease Mutations from Human Orthologs. Int. J. Mol. Sci. 2021, 22, 6200.
- Rzepnikowska, W.; Flis, K.; Muñoz-Braceras, S.; Menezes, R.; Escalante, R.; Zoladek, T. Yeast and other lower eukaryotic organisms for studies of Vps13 proteins in health and disease. Traffic 2017, 18, 711–719.
- Rzepnikowska, W.; Flis, K.; Kaminska, J.; Grynberg, M.; Urbanek, A.; Ayscough, K.R.; Zoladek, T. Amino acid substitution equivalent to human chorea-acanthocytosis I2771R in yeast Vps13 protein affects its binding to phosphatidylinositol 3-phosphate. Hum. Mol. Genet. 2017, 26, 1497–1510.
- Park, J.S.; Thorsness, M.K.; Policastro, R.; McGoldrick, L.L.; Hollingsworth, N.M.; Thorsness, P.E.; Neiman, A.M. Yeast Vps13 promotes mitochondrial function and is localized at membrane contact sites. Mol. Biol. Cell 2016, 27, 2435–2449.
- Soczewka, P.; Kolakowski, D.; Smaczynska-de Rooij, I.; Rzepnikowska, W.; Ayscough, K.R.; Kaminska, J.; Zoladek, T. Yeast-model-based study identified myosin- and calcium-dependent calmodulin signalling as a potential target for drug intervention in chorea-acanthocytosis. Dis. Models Mech. 2019, 12, dmm036830.
- Goodson, H.V.; Anderson, B.L.; Warrick, H.M.; Pon, L.A.; Spudich, J.A. Synthetic lethality screen identifies a novel yeast myosin I gene (MYO5): Myosin I proteins are required for polarization of the actin cytoskeleton. J. Cell Biol. 1996, 133, 1277–1291.
- Geli, M.I.; Riezman, H. Role of type I myosins in receptor-mediated endocytosis in yeast. Science 1996, 272, 533–535.
- Cyert, M.S. Calcineurin signaling in Saccharomyces cerevisiae: How yeast go crazy in response to stress. Biochem. Biophys. Res. Commun. 2003, 311, 1143–1150.
- Cyert, M.S.; Kunisawa, R.; Kaim, D.; Thorner, J. Yeast has homologs (CNA1 and CNA2 gene products) of mammalian calcineurin, a calmodulin-regulated phosphoprotein phosphatase. Proc. Natl. Acad. Sci. USA 1991, 88, 7376–7380.
- Cyert, M.S.; Thorner, J. Regulatory subunit (CNB1 gene product) of yeast Ca2+/calmodulin-dependent phosphoprotein phosphatases is required for adaptation to pheromone. Mol. Cell Biol. 1992, 12, 3460–3469.
- Liu, Y.; Ishii, S.; Tokai, M.; Tsutsumi, H.; Ohki, O.; Akada, R.; Tanaka, K.; Tsuchiya, E.; Fukui, S.; Miyakawa, T. The Saccharomyces cerevisiae genes (CMP1 and CMP2) encoding calmodulin-binding proteins homologous to the catalytic subunit of mammalian protein phosphatase 2B. Mol. Gen. Genet. 1991, 227, 52–59.
- Stathopoulos, A.M.; Cyert, M.S. Calcineurin acts through the CRZ1/TCN1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 1997, 11, 3432–3444.
- Stathopoulos-Gerontides, A.; Guo, J.J.; Cyert, M.S. Yeast calcineurin regulates nuclear localization of the Crz1p transcription factor through dephosphorylation. Genes Dev. 1999, 13, 798–803.
- Caraveo, G.; Soste, M.; Cappelleti, V.; Fanning, S.; van Rossum, D.B.; Whitesell, L.; Huang, Y.; Chung, C.Y.; Baru, V.; Zaichick, S.; et al. FKBP12 contributes to α-synuclein toxicity by regulating the calcineurin-dependent phosphoproteome. Proc. Natl. Acad. Sci. USA 2017, 114, E11313–E11322.
- Wardaszka, P.; Soczewka, P.; Sienko, M.; Zoladek, T.; Kaminska, J. Partial Inhibition of Calcineurin Activity by Rcn2 as a Potential Remedy for Vps13 Deficiency. Int. J. Mol. Sci. 2021, 22, 1193.
- Mehta, S.; Li, H.; Hogan, P.G.; Cunningham, K.W. Domain architecture of the regulators of calcineurin (RCANs) and identification of a divergent RCAN in yeast. Mol. Cell Biol. 2009, 29, 2777–2793.
- Kingsbury, T.J.; Cunningham, K.W. A conserved family of calcineurin regulators. Genes Dev. 2000, 14, 1595–1604.
- Francis, C.E.; Bai, Y. Differential expression of cyclosporine A-Induced calcineurin isoform-specific matrix metalloproteinase 9 (MMP-9) in renal fibroblasts. Biochem. Biophys. Res. Commun. 2018, 503, 2549–2554.
- Usuda, N.; Arai, H.; Sasaki, H.; Hanai, T.; Nagata, T.; Muramatsu, T.; Kincaid, R.L.; Higuchi, S. Differential subcellular localization of neural isoforms of the catalytic subunit of calmodulin-dependent protein phosphatase (calcineurin) in central nervous system neurons: Immunohistochemistry on formalin-fixed paraffin sections employing antigen retrieval by microwave irradiation. J. Histochem. Cytochem. 1996, 44, 13–18.
- Eastwood, S.L.; Salih, T.; Harrison, P.J. Differential expression of calcineurin A subunit mRNA isoforms during rat hippocampal and cerebellar development. Eur. J. Neurosci. 2005, 22, 3017–3024.
- Zhang, Y.; Liu, R.B.; Cao, Q.; Fan, K.Q.; Huang, L.J.; Yu, J.S.; Gao, Z.J.; Huang, T.; Zhong, J.Y.; Mao, X.T.; et al. USP16-mediated deubiquitination of calcineurin A controls peripheral T cell maintenance. J. Clin. Investig. 2019, 129, 2856–2871.
- Wang, L.; Cheng, N.; Wang, P.; Li, J.; Jia, A.; Li, W.; Zhang, N.; Yin, Y.; Tong, L.; Wei, Q.; et al. A novel peptide exerts potent immunosuppression by blocking the two-site interaction of NFAT with calcineurin. J. Biol. Chem. 2020, 295, 2760–2770.
- Noguchi, H.; Sugimoto, K.; Miyagi-Shiohira, C.; Nakashima, Y.; Kobayashi, N.; Saitoh, I.; Watanabe, M.; Noguchi, Y. RCAN-11R peptide provides immunosuppression for fully mismatched islet allografts in mice. Sci. Rep. 2017, 7, 3043.
- Kipanyula, M.J.; Kimaro, W.H.; Seke Etet, P.F. The Emerging Roles of the Calcineurin-Nuclear Factor of Activated T-Lymphocytes Pathway in Nervous System Functions and Diseases. J. Aging Res. 2016, 2016, 5081021.
- Mizuguchi, T.; Nakashima, M.; Kato, M.; Okamoto, N.; Kurahashi, H.; Ekhilevitch, N.; Shiina, M.; Nishimura, G.; Shibata, T.; Matsuo, M.; et al. Loss-of-function and gain-of-function mutations in PPP3CA cause two distinct disorders. Hum. Mol. Genet. 2018, 27, 1421–1433.
- Caraveo, G.; Auluck, P.K.; Whitesell, L.; Chung, C.Y.; Baru, V.; Mosharov, E.V.; Yan, X.; Ben-Johny, M.; Soste, M.; Picotti, P.; et al. Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc. Natl. Acad. Sci. USA 2014, 111, E3544–E3552.
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436.
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111.
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G.J.D.S. The Interplay between Ca2+ Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004.
- Huang, D.X.; Yu, X.; Yu, W.J.; Zhang, X.M.; Liu, C.; Liu, H.P.; Sun, Y.; Jiang, Z.P. Calcium Signaling Regulated by Cellular Membrane Systems and Calcium Homeostasis Perturbed in Alzheimer’s Disease. Front. Cell Dev. Biol. 2022, 10, 834962.
- Catoni, C.; Calì, T.; Brini, M. Calcium, Dopamine and Neuronal Calcium Sensor 1: Their Contribution to Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 55.
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94.
- Marambaud, P.; Dreses-Werringloer, U.; Vingtdeux, V. Calcium signaling in neurodegeneration. Mol. Neurodegener. 2009, 4, 20.
- Pelzl, L.; Elsir, B.; Sahu, I.; Bissinger, R.; Singh, Y.; Sukkar, B.; Honisch, S.; Schoels, L.; Jemaà, M.; Lang, E.; et al. Lithium Sensitivity of Store Operated Ca2+ Entry and Survival of Fibroblasts Isolated from Chorea-Acanthocytosis Patients. Cell Physiol. Biochem. 2017, 42, 2066–2077.
- Pelzl, L.; Hauser, S.; Elsir, B.; Sukkar, B.; Sahu, I.; Singh, Y.; Höflinger, P.; Bissinger, R.; Jemaà, M.; Stournaras, C.; et al. Lithium Sensitive ORAI1 Expression, Store Operated Ca. Sci. Rep. 2017, 7, 6457.
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100.
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139 (Suppl. 1), 179–197.
- Ejaz, H.W.; Wang, W.; Lang, M. Copper Toxicity Links to Pathogenesis of Alzheimer’s Disease and Therapeutics Approaches. Int. J. Mol. Sci. 2020, 21, 7660.
- Gromadzka, G.; Tarnacka, B.; Flaga, A.; Adamczyk, A. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 9259.
- Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Anwer, M.K.; Makeen, H.A.; Albratty, M.; Mohan, S.; Bungau, S. Mechanistic Insights Expatiating the Redox-Active-Metal-Mediated Neuronal Degeneration in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 678.
- Ackerman, C.M.; Chang, C.J. Copper signaling in the brain and beyond. J. Biol. Chem. 2018, 293, 4628–4635.
- Fox, J.H.; Kama, J.A.; Lieberman, G.; Chopra, R.; Dorsey, K.; Chopra, V.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Hersch, S. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS ONE 2007, 2, e334.
- Aaseth, J.; Skalny, A.V.; Roos, P.M.; Alexander, J.; Aschner, M.; Tinkov, A.A. Copper, Iron, Selenium and Lipo-Glycemic Dysmetabolism in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9461.
- Schrag, M.; Mueller, C.; Oyoyo, U.; Smith, M.A.; Kirsch, W.M. Iron, zinc and copper in the Alzheimer’s disease brain: A quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 2011, 94, 296–306.
- Squitti, R.; Faller, P.; Hureau, C.; Granzotto, A.; White, A.R.; Kepp, K.P. Copper Imbalance in Alzheimer’s Disease and Its Link with the Amyloid Hypothesis: Towards a Combined Clinical, Chemical, and Genetic Etiology. J. Alzheimers Dis. 2021, 83, 23–41.
- Li, D.D.; Zhang, W.; Wang, Z.Y.; Zhao, P. Serum Copper, Zinc, and Iron Levels in Patients with Alzheimer’s Disease: A Meta-Analysis of Case-Control Studies. Front. Aging Neurosci. 2017, 9, 300.
- Guan, C.; Dang, R.; Cui, Y.; Liu, L.; Chen, X.; Wang, X.; Zhu, J.; Li, D.; Li, J.; Wang, D. Characterization of plasma metal profiles in Alzheimer’s disease using multivariate statistical analysis. PLoS ONE 2017, 12, e0178271.
- Lee, J.H.; Lee, S.M.; Baik, S.K. Demonstration of striatopallidal iron deposition in chorea-acanthocytosis by susceptibility-weighted imaging. J. Neurol. 2011, 258, 321–322.
- Soczewka, P.; Flis, K.; Tribouillard-Tanvier, D.; di Rago, J.P.; Santos, C.N.; Menezes, R.; Kaminska, J.; Zoladek, T. Flavonoids as Potential Drugs for VPS13-Dependent Rare Neurodegenerative Diseases. Genes 2020, 11, 828.
- Soczewka, P.; Tribouillard-Tanvier, D.; di Rago, J.P.; Zoladek, T.; Kaminska, J. Targeting Copper Homeostasis Improves Functioning of vps13Δ Yeast Mutant Cells, a Model of VPS13-Related Diseases. Int. J. Mol. Sci. 2021, 22, 2248.
- de Andrade Teles, R.B.; Diniz, T.C.; Costa Pinto, T.C.; de Oliveira Júnior, R.G.; Gama E Silva, M.; de Lavor, É.; Fernandes, A.W.C.; de Oliveira, A.P.; de Almeida Ribeiro, F.P.R.; da Silva, A.A.M.; et al. Flavonoids as Therapeutic Agents in Alzheimer’s and Parkinson’s Diseases: A Systematic Review of Preclinical Evidences. Oxid. Med. Cell. Longev. 2018, 2018, 7043213.
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750.
- Rodríguez-García, C.; Sánchez-Quesada, C.; Toledo, E.; Delgado-Rodríguez, M.; Gaforio, J.J. Naturally Lignan-Rich Foods: A Dietary Tool for Health Promotion? Molecules 2019, 24, 917.
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Orhan, I.E.; Daglia, M.; Manayi, A.; Gortzi, O.; Nabavi, S.M. Neuroprotective effects of chrysin: From chemistry to medicine. Neurochem. Int. 2015, 90, 224–231.
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47.
- Antonini, A.; Abbruzzese, G.; Barone, P.; Bonuccelli, U.; Lopiano, L.; Onofrj, M.; Zappia, M.; Quattrone, A. COMT inhibition with tolcapone in the treatment algorithm of patients with Parkinson’s disease (PD): Relevance for motor and non-motor features. Neuropsychiatr. Dis. Treat. 2008, 4, 1–9.
- Willman, C.; Tadi, P. StatPearls. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560593/ (accessed on 29 April 2022).
- Bors, W.; Heller, W.; Michel, C.; Saran, M. Flavonoids as antioxidants: Determination of radical-scavenging efficiencies. Methods Enzymol. 1990, 186, 343–355.
- Fernandez, M.T.; Mira, M.L.; Florêncio, M.H.; Jennings, K.R. Iron and copper chelation by flavonoids: An electrospray mass spectrometry study. J. Inorg. Biochem. 2002, 92, 105–111.
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol. Med. 1996, 20, 933–956.
- Ruotolo, R.; Marchini, G.; Ottonello, S. Membrane transporters and protein traffic networks differentially affecting metal tolerance: A genomic phenotyping study in yeast. Genome Biol. 2008, 9, R67.
- Jiang, L.; Cao, C.; Zhang, L.; Lin, W.; Xia, J.; Xu, H.; Zhang, Y. Cadmium-induced activation of high osmolarity glycerol pathway through its Sln1 branch is dependent on the MAP kinase kinase kinase Ssk2, but not its paralog Ssk22, in budding yeast. FEMS Yeast Res. 2014, 14, 1263–1272.
- Dix, D.R.; Bridgham, J.T.; Broderius, M.A.; Byersdorfer, C.A.; Eide, D.J. The FET4 gene encodes the low affinity Fe(II) transport protein of Saccharomyces cerevisiae. J. Biol. Chem. 1994, 269, 26092–26099.
- Hechtberger, P.; Zinser, E.; Saf, R.; Hummel, K.; Paltauf, F.; Daum, G. Characterization, quantification and subcellular localization of inositol-containing sphingolipids of the yeast, Saccharomyces cerevisiae. Eur. J. Biochem. 1994, 225, 641–649.
- Kraft, M.L. Sphingolipid Organization in the Plasma Membrane and the Mechanisms That Influence It. Front. Cell Dev. Biol. 2016, 4, 154.
- Goins, L.; Spassieva, S. Sphingoid bases and their involvement in neurodegenerative diseases. Adv. Biol. Regul. 2018, 70, 65–73.
- López-García, B.; Gandía, M.; Muñoz, A.; Carmona, L.; Marcos, J.F. A genomic approach highlights common and diverse effects and determinants of susceptibility on the yeast Saccharomyces cerevisiae exposed to distinct antimicrobial peptides. BMC Microbiol. 2010, 10, 289.
- Kaniak-Golik, A.; Skoneczna, A. Mitochondria-nucleus network for genome stability. Free Radic Biol. Med. 2015, 82, 73–104.
- Abdel Hadi, L.; Di Vito, C.; Marfia, G.; Ferraretto, A.; Tringali, C.; Viani, P.; Riboni, L. Sphingosine Kinase 2 and Ceramide Transport as Key Targets of the Natural Flavonoid Luteolin to Induce Apoptosis in Colon Cancer Cells. PLoS ONE 2015, 10, e0143384.
- Pena, M.M.; Puig, S.; Thiele, D.J. Characterization of the Saccharomyces cerevisiae high affinity copper transporter Ctr3. J. Biol. Chem. 2000, 275, 33244–33251.
- Dancis, A.; Yuan, D.S.; Haile, D.; Askwith, C.; Eide, D.; Moehle, C.; Kaplan, J.; Klausner, R.D. Molecular characterization of a copper transport protein in S. cerevisiae: An unexpected role for copper in iron transport. Cell 1994, 76, 393–402.
- Dancis, A.; Haile, D.; Yuan, D.S.; Klausner, R.D. The Saccharomyces cerevisiae copper transport protein (Ctr1p). Biochemical characterization, regulation by copper, and physiologic role in copper uptake. J. Biol. Chem. 1994, 269, 25660–25667.
- Li, H.; Wang, J.; Wu, C.; Wang, L.; Chen, Z.S.; Cui, W. The combination of disulfiram and copper for cancer treatment. Drug Discov. Today 2020, 25, 1099–1108.
- Kirshner, J.R.; He, S.; Balasubramanyam, V.; Kepros, J.; Yang, C.Y.; Zhang, M.; Du, Z.; Barsoum, J.; Bertin, J. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol. Cancer Ther. 2008, 7, 2319–2327.
- Wangpaichitr, M.; Wu, C.; You, M.; Maher, J.C.; Dinh, V.; Feun, L.G.; Savaraj, N. N′,N′-Dimethyl-N′,N′-bis(phenylcarbonothioyl) Propanedihydrazide (Elesclomol) Selectively Kills Cisplatin Resistant Lung Cancer Cells through Reactive Oxygen Species (ROS). Cancers 2009, 1, 23–38.
- Reeder, N.L.; Kaplan, J.; Xu, J.; Youngquist, R.S.; Wallace, J.; Hu, P.; Juhlin, K.D.; Schwartz, J.R.; Grant, R.A.; Fieno, A.; et al. Zinc pyrithione inhibits yeast growth through copper influx and inactivation of iron-sulfur proteins. Antimicrob. Agents Chemother. 2011, 55, 5753–5760.
- Yuan, D.S.; Dancis, A.; Klausner, R.D. Restriction of copper export in Saccharomyces cerevisiae to a late Golgi or post-Golgi compartment in the secretory pathway. J. Biol. Chem. 1997, 272, 25787–25793.
- Yuan, D.S.; Stearman, R.; Dancis, A.; Dunn, T.; Beeler, T.; Klausner, R.D. The Menkes/Wilson disease gene homologue in yeast provides copper to a ceruloplasmin-like oxidase required for iron uptake. Proc. Natl. Acad. Sci. USA 1995, 92, 2632–2636.
- Stearman, R.; Yuan, D.S.; Yamaguchi-Iwai, Y.; Klausner, R.D.; Dancis, A. A permease-oxidase complex involved in high-affinity iron uptake in yeast. Science 1996, 271, 1552–1557.
- Askwith, C.; Eide, D.; Van Ho, A.; Bernard, P.S.; Li, L.; Davis-Kaplan, S.; Sipe, D.M.; Kaplan, J. The FET3 gene of S. cerevisiae encodes a multicopper oxidase required for ferrous iron uptake. Cell 1994, 76, 403–410.
- Gaxiola, R.A.; Yuan, D.S.; Klausner, R.D.; Fink, G.R. The yeast CLC chloride channel functions in cation homeostasis. Proc. Natl. Acad. Sci. USA 1998, 95, 4046–4050.
- Soma, S.; Latimer, A.J.; Chun, H.; Vicary, A.C.; Timbalia, S.A.; Boulet, A.; Rahn, J.J.; Chan, S.S.L.; Leary, S.C.; Kim, B.E.; et al. Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, 8161–8166.
- Guthrie, L.M.; Soma, S.; Yuan, S.; Silva, A.; Zulkifli, M.; Snavely, T.C.; Greene, H.F.; Nunez, E.; Lynch, B.; De Ville, C.; et al. Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science 2020, 368, 620–625.

