2. Calcium Signalling as a Potential Target for Drug Intervention in VPS13-Dependent Neurodegenerative Diseases
The knowledge about VPS13 proteins is still not sufficient to develop a specific therapy for patients. To date, gene therapy is also not available. Only symptomatic treatment is used, and new methods to alleviate the symptoms are needed. The most efforts for treatment development are reported for ChAc. The deep brain stimulation is used to relieve movement problems
[10][118], botulinum toxin to treat dystonia and selected drugs for seizure control
[11][77]. Based on the finding that Lyn kinase is hyperactivated in red blood cells of ChAc patients and neurons
[12][13][89,90], the inhibitors of this kinase were also tested. As Lyn kinase inhibition represents a potential treatment in ChAc to restore some neuronal function
[12][89], the trial to experimentally treat 3 patients was conducted. Although a partial restoration of the actin cytoskeleton was observed in red blood cells, the lack of improvement of neurological symptoms was noted after six-month drug administration
[14][123]. Further, studies on mice models revealed that the drug tested, dasatinib, is not able to cross the blood–brain barrier, but another potential kinase inhibitor, nilotinib, can do that and improve not only haematological but also neurological defects in mice
[15][16][124,125]. Since the variety of potential treatments is very limited, there is a great need for new drug candidates which could be effective in ChAc patients.
To find therapeutic alternatives based on the understanding of cell biology, using model organisms is necessary. Several yeast models of
VPS13-dependent diseases were constructed, which show various phenotypes
[17][18][19][20][21][10,11,13,48,51]. However, these phenotypes were not suitable for high-throughput screens. Finding novel growth phenotype of
vps13, hypersensitivity to commonly used detergent sodium dodecyl sulfate (SDS), enable suppressor screens
[22][126]. This gave better insight into the pathways which are disturbed in these cells which allowed for finding ways to overcome the observed defects.
Specifically, SDS hypersensitivity growth phenotype was used in a screen for multicopy suppressors of
vps13-I2749R mutation, and several plasmids responsible for improved growth were isolated and analysed. One of them contained a fragment of the
MYO3 gene (
MYO3-N) encoding N-terminal part (amino acid residues (aa) 1–775) of Myo3 protein (Myo3-N)
[22][126]. Myo3 is a type I myosin protein involved in actin cytoskeleton organisation and endocytosis
[23][24][127,128].
MYO3-N overexpression was also improving growth of the
vps13Δ mutant in the presence of SDS. Importantly, the suppression by
MYO3-N was not limited to SDS-hypersensitivity.
MYO3-N corrected two other
vps13Δ defects: depolarisation of the actin cytoskeleton and hypersensitivity to canavanine, the phenotypes implying defective endocytosis. Indeed, endocytic reporters (Las17, Myo3, Myo5 and Abp1 tagged with fluorescent proteins) that localise to sites of endocytosis, were shown to be present longer on the plasma membrane in the
vps13Δ cells than in the wild-type, indicating delayed endocytosis.
MYO3-N overexpression shortened patch lifetimes for Las17, Myo3 and Myo5. To conclude
MYO3-N overexpression could partially improve endocytosis in
vps13Δ alleviating canavanine hypersensitivity
[22][126].
In further analysis, the mechanism of
MYO3-N action was elucidated. The suppressing
MYO3-N fragment encodes myosin motor domain and a linker with two calmodulin binding motifs (IQ1 and IQ2). It is possible that suppression could be achieved by binding with calmodulin (Cmd1)—a conserved, calcium-binding protein mediating calcium signalling in cell
[25][129]. To test this possibility, mutations disrupting both IQ motifs were introduced into
MYO3-N and resulted
myo3-iq1/2 was not able to suppress
vps13Δ and the interaction between Myo3-iq1/2 and Cmd1 was abolished. These results indicated that binding of calmodulin to Myo3-N is necessary for
MYO3-N-based suppression of
vps13Δ
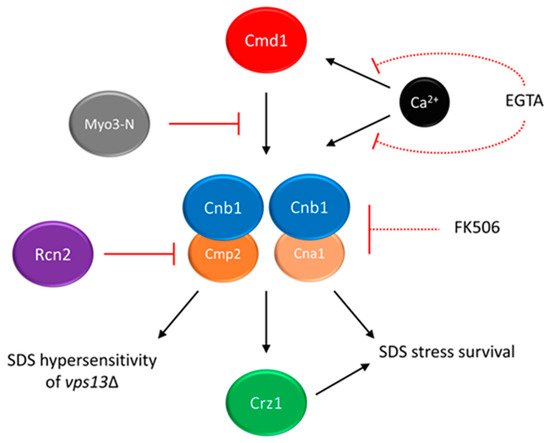
[22][126]. The hypothesis that titrating of calmodulin by Myo3-N may result in lowering the activity of one of its downstream targets, calcineurin, was formulated. In yeast, calcineurin consists of one regulatory (Cnb1) and one of the two catalytic (Cna1 and Cmp2) subunits
[26][27][28][130,131,132] (see
Figure 1). In response to changes in calcium concentration, calcineurin regulates gene expression via its target, the Crz1 transcription factor
[29][30][133,134]. In fact calcineurin activity is higher in the
vps13Δ cells than in the wild-type, and
MYO3-N overexpression reduces it (
Figure 1)
[22][126]. Remarkably, the Cnb1 subunit, which is essential for calcineurin activity, is required for
MYO3-N-based suppression of
vps13Δ. This indicates that the suppression mechanism of
MYO3-N relies on downregulating calcineurin activity. However, calcineurin must not be shut off completely (
Figure 1). This result resembles a study which showed that in a yeast model of PD, moderate calcineurin inhibition reduces α-synuclein toxicity, and deletion or overexpression of genes encoding calcineurin subunits exacerbate it
[31][135].
Figure 1. The calmodulin–calcineurin pathway and its impact on yeast growth in the presence of SDS stress. Partial inhibition of calcineurin (Cnb1 + Cna1/Cmp2) activity suppresses the SDS-hypersensitivity of
vps13Δ cells. Multicopy suppressors
MYO3-N and
RCN2 act by titrating calmodulin or blocking the activity of Cmp2 catalytic subunit, respectively. Chemical suppressors EGTA and FK-506 act by chelating the calcium ions required for calmodulin and calcineurin activities or inhibiting total calcineurin activity, respectively. As full inhibition of calcineurin activity is toxic in the presence of SDS stress, both chemical suppressors must be used in moderate concentration, indicated by dashed lines. Figure is based on results presented in Soczewka et al. 2019 and Wardaszka et al. 2021
[22][32][126,136].
The identification of the
RCN2 gene as another suppressor of
vps13Δ
[32][136] is in line with the results described above.
RCN2 encodes Rcn2 protein, a negative calcineurin regulator
[33][34][137,138]. Interestingly, Rcn2 binds preferentially to the Cmp2 calcineurin catalytic subunit in comparison to the Cna1 catalytic subunit. Analysis indicated that the N-terminal fragment of Rcn2 is necessary to reduce calcineurin activity, maintain the Rcn2 interaction with Cmp2 and to improve growth of
vps13Δ (
Figure 1). Moreover, the deletion of
CMP2 actually suppresses
vps13Δ, contrary to
CNA1 and previously shown
CNB1 deletions, which negatively influence
vps13Δ growth in the presence of SDS. The
CNA1 and
CNB1 are also required for
vps13Δ suppression by
RCN2 [32][136] as for
MYO3-N, indicating that
vps13Δ suppression is achieved by reducing calcineurin activity related to Cmp2 catalytic subunit (
Figure 1), while the activity mediated by Cna1 is crucial for SDS stress survival. Similarly, in mammals catalytic calcineurin subunits are differently expressed and regulated
[35][36][37][38][139,140,141,142]. Knowledge about the specificity of various calcineurin forms and their role in disease molecular pathologies may contribute to development of novel and specific calcineurin inhibitors. Such specific peptide inhibitors of calcineurin would allow calcineurin activity involved in a disease to be downregulated without disturbing calcineurin-related processes required for healthy organism functioning. Various peptide inhibitors of human calcineurin were already studied
[39][40][143,144]. This approach could, at least partially, eliminate side effects of general calcineurin inhibitors, such as FK-506 immunosuppressant and cyclosporin A.
As partial reduction of calcineurin activity was responsible for the suppression, it is possible that
vps13Δ SDS-hypersensitivity could be mitigated by pharmacological calcineurin inhibition. Indeed ethylene glycol tetraacetic acid (EGTA), which limits the calcium availability required for calmodulin and calcineurin activity, and FK-506 (
Figure 1) improved the
vps13Δ growth, showing that it is possible to achieve
vps13Δ suppression in a pharmacological manner
[22][126]. Moreover, FK-506 was active only in very low concentration, and higher concentration caused toxicity. This is in line with genetic experiments and shows that basal calcineurin activity is required for SDS protection.
The proper functioning of calcium signalling is crucial for cells. Especially in neurons, the concentration and storage of Ca
2+ ions in specific compartments are precisely controlled. Dysregulation of Ca
2+ signalling is observed in ageing neurons and neurons affected by neurodegenerative diseases such as Alzheimer's disease (AD), Parkinson's disease (PD) and Huntington disease (HD). Based on these findings, a hypothesis was formed that the dysregulation of Ca
2+ signalling is the primary basis for the pathogenesis of neurodegenerative diseases. Indeed the changes in activity of calcineurin were observed in several neurological diseases
[41][42][145,146], and its increased activity was reported in yeast models of PD
[43][147]. Moreover, the store-operated calcium entry (SOCE), a mechanism of acquiring extracellular calcium triggered by Ca
2+ depletion in the ER
[44][148], was found defective in all major neurodegenerative diseases including AD, PD and HD
[45][149]. The nature of impairment is characteristic for each of these diseases. While HD and PD are characterised by an excessive depletion of Ca
2+ from ER stores in neurons, in AD neurons the ER is overloaded with Ca
2+, as described in several recent reviews
[45][46][47][48][49][50][149,150,151,152,153,154]. In addition, in ChAc patient-derived fibroblasts, levels of SOCE components, ORAI1 and STIM1 were reduced, and SOCE activity was downregulated
[51][155]. Similar defects were observed in neurons generated from ChAc patient-derived induced pluripotent stem cells (iPSC), and these alteration in SOCE functioning, at least partially, contributed to neurodegeneration
[52][156]. Despite enormous efforts, AD, PD and HD are still incurable, and only symptomatic relief drugs are available; research on effective treatment is still ongoing. One line of search for new therapeutic treatment is based on the Ca
2+ signalling hypothesis of neurodegeneration. The effect of modulating the release of Ca
2+ ions from ER storage sites and transport to cells was studied
[53][157]. The relevance of Ca
2+ signalling for neurodegenerative diseases further supports research focused on investigating it as a potential therapeutic target in ChAc patients.
3. Copper and Iron Homeostasis as a Potential Target for Treatment of VPS13-Dependent Neurodegenerative Diseases
Iron and copper are linked with neurodegenerative diseases with protein aggregation. AD, PD and HP are characterised by increased iron and/or copper levels in specific brain regions that are accompanied by cellular damage and oxidative stress
[54][55][56][57][158,159,160,161]. Iron and copper interact with the amyloid precursor protein (APP) and its peptide derivative, amyloid beta (Aβ), both of which are involved in AD, and with α-synuclein which is involved in PD, while copper binds to huntingtin involved in HD
[58][59][162,163]. It has been suggested that this interaction mediates protein aggregation and contributes to disease development
[54][158]. Moreover, copper and iron stimulate the formation of advanced glycation end-products (AGEs), which are toxic and induce aggregation of proteins including those associated with the pathogenesis of AD
[60][164]. However, a meta-analysis indicates a copper deficiency in the brain of AD cases
[61][165], and most meta-analyses results suggest that overall and unbound copper are present in higher concentrations in serum samples of AD patients
[62][63][64][166,167,168], suggesting copper dyshomeostasis. Based on these findings various metal chelators are under study for AD and PD in mouse models and in clinical trials
[54][55][56][158,159,160].
There are not many reports on iron or copper dyshomeostasis in
VPS13-dependent diseases or metal ion contribution in their pathogenesis so far
[65][169]. Finding
FET4, encoding iron transporter, and
CTR3, encoding copper transporter, as suppressors of
vps13Δ
[66][67][41,170] provides a hint that metal disturbances could contribute to the pathogenesis of
VPS13-dependent diseases and opens the possibility of discovering new drugs and drug targets aimed at the normalisation of these disturbances. For this purpose, a chemical suppressor screen of compounds from the Prestwick Chemical Library, a collection of 1280 drugs (most of which have been approved for use in humans), was performed and resulted in the identification of luteolin and tolcapone as
vps13Δ chemical suppressors
[66][41]. Luteolin is a natural compound belonging to the class of polyphenols called flavonoids which are plant secondary metabolites. They are associated with antioxidant, antiviral, antibacterial, anticancer and neuroprotective activities, and their therapeutic potential has been extensively studied
[68][69][70][71][171,172,173,174]. The core of flavonoids is formed by two benzene rings connected with a heterocyclic pyranic ring. Their physico-chemical properties are determined by functional groups and their location in the flavonoid core
[69][72][172,175]. Tolcapone, which is a drug used in the treatment of PD
[73][176], has some structural similarities with luteolin, such as the location of benzene rings and hydroxyl groups on adjacent carbons. Moreover, both these drugs showed comparable activities when used in the same concentrations. These features indicate that luteolin and tolcapone could have the same mechanism of action. Tolcapone, however, exhibits serious adverse effects
[74][177], while flavonoids are generally safe. The Prestwick Chemical Library contained one more flavonoid, ipriflavone, but it did not overcome the
vps13Δ growth defect. To find out more about flavonoids as suppressors, a follow-up screen of the in-house library of approximately 50 natural compounds was performed
[66][41]. An additional five drugs were identified as
vps13Δ suppressors in this screen. Four of them—quercetin, pentaacetylquercetin, myricetin and fisetin—are flavonoids. The fifth compound, corilagin, belongs to tannins. In the collection of tested compounds, another flavonoid, kaempferol, was present but it did not improve
vps13Δ growth. The only structural difference that distinguishes kaempferol from active flavonoids was that in the kaempferol structure, none of the hydroxyl groups are bound to adjacent carbon atoms. This criterion required for
vps13Δ suppression by flavonoids was further confirmed in the structure–activity–relationship (SAR) analysis. The other structural criteria established during SAR analysis implicated that the heterocyclic pyranic ring must contain a double bond between C2 and C3 atoms and a carbonyl group, yet the ring itself does not necessarily have to be closed. A similar case is tolcapone, where the criteria regarding hydroxyl and carbonyl groups are met and benzyl rings are not connected by heterocyclic pyranic ring but by a carbonyl group.
The structural criteria required for
vps13Δ suppression overlaps with those previously described for flavonoids responsible for the antioxidant and metal chelation properties
[75][76][77][178,179,180]. As
vps13Δ was shown to be hypersensitive to cadmium
[78][79][181,182], a heavy metal that causes oxidative stress, and all of the flavonoids that suppressed
vps13Δ SDS hypersensitivity were also active when tested for
vps13Δ cadmium hypersensitivity, a possible mechanism of their action could rely on their antioxidant properties. Contrary to this prediction, some of the tested flavonoids, such as kaempferol, did not improve
vps13Δ growth, despite having higher antioxidant potential than the suppressing flavonoids
[77][180]. This contradiction suggests that the alleviation of oxidative stress is a rather unlikely mechanism for flavonoid suppression of
vps13Δ. Thus, it was hypothesised that metal chelation properties could possibly be important for
vps13Δ suppression. This was supported by the fact, that one of the identified genes in the multicopy suppressor screen was
FET4 [66][41], which encodes a plasma membrane low-affinity iron transporter
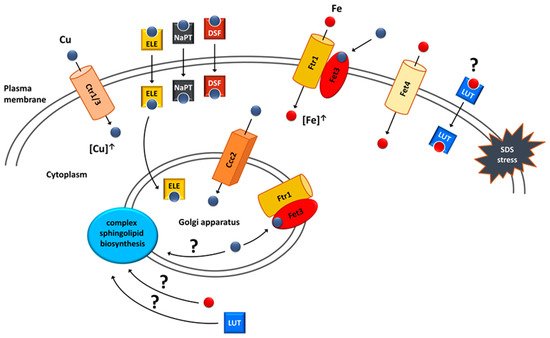
[80][183] (
Figure 2). In addition, iron salts alone improved
vps13Δ growth. These results highlight the importance of iron in the protection against SDS stress. The potential interaction between luteolin and
FET4 overexpression was tested and luteolin improved
vps13Δ growth regardless of
FET4 overexpression, and no additive effects were observed
[66][41]. This indicates that luteolin may involve iron in its mechanism and could act on the same process as
FET4 (
Figure 2). It remains to be tested directly whether luteolin acts by compensating iron deficiency in
vps13Δ cells or by influencing other cellular process.
Figure 25. Suppression mechanisms of
vps13Δ SDS hypersensitivity that involve copper or iron homeostasis. Suppression could be achieved by (I) overexpression of genes encoding copper transporters Ctr1, Ctr3 and Ccc2; (II) overexpression of the
FET4 gene encoding the Fet4 iron transporter; (III) treatment with copper ionophores (ELE—elesclomol; NaPT—sodium pyrithione; DSF—disulfiram); (IV) treatment with luteolin (LUT). Internalised copper is targeted to the Golgi apparatus where it is incorporated into the Fet3 oxidase, a part of the complex enabling high-affinity iron uptake. This complex is targeted to the plasma membrane via vesicular trafficking and increases the intracellular iron pool used for SDS protection. Extracellular copper can also be directly incorporated into Fet3 oxidase localised in the plasma membrane. It is possible that luteolin could act as iron ionophore. A potential target of copper, iron and luteolin action could be the biosynthesis of complex sphingolipids, which are important membrane components during SDS stress. Figure from Soczewka et al. (2021)
[67][170] was supplemented and modified.
In line with these predictions, possible links between luteolin, iron and sphingolipids (
Figure 2) were tested
[66][41]. Sphingolipids are structural components of membranes with the highest enrichment in the plasma membrane, and they are involved in signalling and regulatory processes in cells
[81][82][184,185]. They are especially abundant in cells of the nervous system, and alterations in their metabolism are implicated in neurodegenerative diseases such as multiple sclerosis and Sandhoff disease
[83][186]. Previous studies showed that the
IPT1 gene, which encodes the inositol-phosphotransferase crucial for sphingolipid biosynthesis, is important for the SDS stress response in yeast
[84][187]. Moreover, iron serves as a cofactor in enzymes of sphingolipid biosynthesis pathway
[85][188], and luteolin was shown to increase ceramide level, one of the key sphingolipid, in cancer cell line
[86][189]. This raised the hypothesis that both iron and luteolin could act on the sphingolipid biosynthesis pathway to suppress
vps13Δ. In agreement with this, the
csg2Δ strain, devoid of another enzyme from the sphingolipid biosynthesis pathway, was also hypersensitive to SDS. Moreover, deletions of both
ipt1Δ and
csg2Δ negatively interacted with
vps13Δ, and luteolin and iron were able to suppress both of the double deletion strains
[66][41]. The contribution of the sphingolipid biosynthesis pathway in
vps13Δ phenotypes and the pathogenesis of
VPS13-dependent diseases requires further study.
The primary finding that points to copper homeostasis as a potential suppression target was the identification of the
CTR3 gene in the screen for multicopy suppressors of
vps13-I2749R and
vps13Δ
[67][170].
CTR3 encodes the plasma membrane copper transporter
[87][190]. Copper relevance for suppression was further confirmed by showing that overexpression of the
CTR1 gene, encoding the main plasma membrane copper transporter
[88][89][191,192], as well as treatment with copper salts improved the
vps13Δ growth (
Figure 2). Moreover, one of the identified compounds as the chemical suppressor was disulfiram
[67][170]—a copper ionophore used for alcoholism treatment, which is gaining interest as a potential anticancer drug
[90][193]. Two additional copper ionophores tested also suppressed
vps13Δ growth defect. One was elesclomol, a candidate anticancer drug
[91][92][194,195], and the other was sodium pyrithione (
Figure 2), which has an anion that is used as an antimicrobial agent
[93][196]. The fact that identified multicopy and chemical suppressors are directly related to copper strongly suggests that increasing the cellular copper concentration could be one of the mechanisms of
vps13Δ suppression. Copper-based suppression was not limited only to the SDS hypersensitivity phenotype.
CTR1 overexpression, as well as treatment with all three copper ionophores, mitigated
vps13Δ growth defect in the presence of cadmium. When testing the suppressors for the canavanine hypersensitivity of
vps13Δ,
CTR1 overexpression only slightly improved
vps13Δ growth, whereas elesclomol was the only active drug. This finding was quite intriguing, because despite the fact that all of the suppressors were copper-related, their mechanisms of action in the cell could be different.
When elucidating potential mechanisms of copper action, the potential copper deficiency and its compensation by improved copper uptake was tested
[67][170]. However, copper measurements and growth tests in the presence of copper chelator did not indicate any copper deficiency in
vps13Δ. Moreover, in copper-abundant conditions, the copper level in
vps13Δ cells was even slightly elevated compared to the wild type. Thus, SDS hypersensitivity of
vps13Δ is not caused by copper deficiency, and suppression of this
vps13Δ does not rely on compensating low copper level; thus, another mechanism of action must exist.
The mechanism standing behind copper-based suppression was related to iron uptake, and its importance for SDS protection was described above
[66][41]. Copper is required for functioning of the high-affinity iron uptake system. Upon internalisation, copper is transported to the Golgi apparatus by Ccc2 ATPase
[94][95][197,198], where it is incorporated into Fet3 oxidase, which, together with Ftr1 permease, forms a complex responsible for the high-affinity iron uptake
[96][97][199,200]. After binding copper, this complex is transported to the plasma membrane, where it enables iron uptake. Increasing intracellular copper pool by
CTR1 overexpression would improve functioning of this system, resulting in increased iron content in the cell. Using a genetic approach, it was shown that this suppression mechanism is possible.
CTR1 did not suppress
vps13Δ when
CCC2 or
FET3 were absent, which indicates that components important for the high-affinity iron uptake system are required for
CTR1-based suppression
[67][170]. To further highlight the relevance of the high-affinity iron uptake system, it was presented that
CCC2 acts as a
vps13Δ multicopy suppressor; therefore, suppression could be achieved by increasing copper transport to the Golgi apparatus. Importantly,
CCC2 overexpression was not effective in the absence of
FET3, which further shows that the high-affinity iron uptake system is essential for copper-based suppression.
The findings from genetic experiments pointing at iron uptake as targets of copper-related suppression were further supported by measurements of iron levels in yeast cells. The results show that the iron level in
vps13Δ was lower than in the wild type, and it increased upon
CTR1 and, to some extent,
CCC2 overexpression
[67][170]. The iron level in
vps13Δ, however, was only moderately decreased in comparison to iron levels observed in the
fet3Δ mutant. It was shown that the
fet3Δ strain was less sensitive to SDS than
vps13Δ
[66][41]; therefore, lower iron levels could contribute to
vps13Δ SDS hypersensitivity only to a low extent. Supplementing media with copper salts greatly increased intracellular iron levels in both wild-type and
vps13Δ strains, which is another indication that increasing the intracellular copper pool increases iron uptake via the Fet3–Ftr1 complex
[67][170]. Interestingly, upon copper supplementation, the difference in iron levels between the wild-type and
vps13Δ strain was higher in comparison to the measurements for yeast cultivated without copper supplementation. This indicates that in the
vps13Δ mutant, copper may not be utilised by the high-affinity iron uptake system as effectively as in the wild type.
It was also tested whether copper ionophores and copper, itself, act via the high-affinity iron uptake system. Neither the tested ionophore nor copper sulphate were active in
fet3Δ
vps13Δ, showing that their mechanism of action relies on iron uptake by the Fet3–Ftr1 complex. Interestingly, however, copper sulphate and elesclomol but neither disulfiram nor sodium pyrithione were able to improve the growth of the
ccc2Δ
vps13Δ double deletion mutant when higher concentrations were applied. Copper sulphate could rescue
ccc2Δ
vps13Δ by the direct incorporation of copper to Fet3 on the plasma membrane
[67][170] (
Figure 2). In this mechanism, there is no need for Ccc2, which delivers copper to the Golgi apparatus where copper binding to Fet3 occurs. In agreement with this, other works showed that the effects of
CCC2 deletion, but not
FET3 deletion, could be overcome by copper supplementation
[94][95][96][98][197,198,199,201]. In the case of elesclomol, it was proposed that the lack of
CCC2 is overcome by its ability to effectively transport copper to the Golgi apparatus
[67][170]. This is supported by previous studies in which elesclomol was able to correct defects observed in the yeast
ccc2Δ mutant, and it was also effective in a Menkes disease model in which the
ATP7A gene, a homologue of yeast
CCC2, is mutated
[99][100][202,203]. It is worth noting that elesclomol was the only ionophore that reduced
vps13Δ hypersensitivity to canavanine, suggesting that its action is broader than the other tested copper ionophores.
Newly identified potential repurposable drugs which are effective in alleviating defects of yeast mutant cells, such as calcineurin inhibitors, flavonoids and copper ionophores, require intensive studies using available human cell and mouse models.