Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Jiang, X.; , .; Meng, L.; Xin, Y. Hypoxic Cells for Tumor Radiotherapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/22829 (accessed on 30 June 2026).
Jiang X, , Meng L, Xin Y. Hypoxic Cells for Tumor Radiotherapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/22829. Accessed June 30, 2026.
Jiang, Xin, , Lingbin Meng, Ying Xin. "Hypoxic Cells for Tumor Radiotherapy" Encyclopedia, https://encyclopedia.pub/entry/22829 (accessed June 30, 2026).
Jiang, X., , ., Meng, L., & Xin, Y. (2022, May 11). Hypoxic Cells for Tumor Radiotherapy. In Encyclopedia. https://encyclopedia.pub/entry/22829
Jiang, Xin, et al. "Hypoxic Cells for Tumor Radiotherapy." Encyclopedia. Web. 11 May, 2022.
Copy Citation
Radiation therapy plays an increasingly important role in cancer treatment. It can inhibit the progression of various cancers through radiation-induced DNA breakage and reactive oxygen species (ROS) overload. Unfortunately, solid tumors, such as breast and lung cancer, often develop a hypoxic microenvironment due to insufficient blood supply and rapid tumor proliferation, thereby affecting the effectiveness of radiation therapy. Restraining hypoxia and improving the curative effect of radiotherapy have become difficult problems. Ferroptosis is a new type of cell death caused by lipid peroxidation due to iron metabolism disorders and ROS accumulation.
hypoxia
ferroptosis
reactive oxygen species (ROS)
1. Introduction
Cancer is one of the most harmful diseases to human health. Due to the progress of treatment technology, the popularization of early cancer screening and other modalities has led to a year-by-year decline in cancer mortality. The latest statistics show a 31% decrease in total cancer deaths between 1991 and 2018 [1]. Radiotherapy (RT) can inhibit the progression of various cancers through radiation-induced DNA breakage and reactive oxygen species (ROS) overload [2]. Unfortunately, solid tumors such as breast cancer and lung cancer often develop a hypoxic microenvironment due to insufficient blood supply and rapid tumor proliferation, thereby affecting the effectiveness of RT. Radiation resistance caused by hypoxia often fails to achieve the expected outcome in the treatment of solid tumors [3]. Overcoming resistance caused by hypoxia has always been a concern for the general population.
Ferroptosis is a new type of cell death caused by lipid peroxidation due to iron metabolism disorders and ROS accumulation. It is different from other regulated cell death (RCD), such as apoptosis, autophagy and pyroptosis, in morphology and mechanisms. Morphologically, ferroptosis is neither featured with typical apoptotic features, such as chromatin agglutination and apoptotic body formation, nor the formation of autophagic vacuoles, unique autophagy features or the release of proinflammatory factors, which is a typical feature of pyroptosis; instead, ferroptotic cells mainly manifest as shrunken mitochondria with increased membrane density and disappeared mitochondrial cristae [4][5]. Mechanistically, although other RCDs can also be triggered by ROS overload [6], ferroptosis is always accompanied by significant iron accumulation, glutathione (GSH) depletion, and changes in genes related to iron homeostasis and lipid metabolism [7]. Polyunsaturated fatty acid (PUFA) is the source of ferroptosis. PUFAs participate in the synthesis of membrane phospholipids catalyzed by acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) [8][9], then cytochrome P450 oxidoreductase (POR) and lipoxygenases (LOX) will oxidize polyunsaturated phospholipids (PUFA-PL) to lipid peroxides (L-OOH) [10][11][12]. Normally, on the one hand, the cytotoxic L-OOH can be reduced to the corresponding alcohols by glutathione peroxidase (GPX4) [13]. On the other hand, it can be converted into L-OO. through Fenton reaction, while L-OO. can be reduced by ubiquinol (CoQH2) to protect cells from lipid peroxidation [14][15]. However, when Fe2+ or PUFA is overloaded or the antioxidant system is imbalanced, L-OO. will accumulate in large quantities, resulting in lipid peroxidation of membrane phospholipids, and ultimately lead to ferroptosis [7]. Cystine/glutamate antiporter solute carrier family 7 member 11 (SLC7A11; also known as xCT) is a key factor regulating ferroptosis. It controls the synthesis of GSH, which allows GPX4 to reduce L-OOH [16]. Furthermore, tetrahydrobiopterin (BH4) and ferroptosis suppressor protein 1 (FSP1) also play an important role in inhibiting ferroptosis by producing CoQH2 [14][17]. In addition, with the increasing research on ferroptosis, it has been found that it plays an important role in the process of cell death caused by radiation. Lang et al. found that radiation can activate the ataxia-telangiectasia mutated gene (ATM) and inhibit the expression of SLC7A11, thereby promoting ferroptosis [18]. Moreover, Lei et al. verified this view and found that ionizing radiation (IR) significantly increased the staining of C11-BODIPY and 4-hydroxynonenal (4-HNE) in tumor cells, thus causing typical morphological changes in ferroptosis [19]. Further studies have shown that ferroptosis inducers such as sulfasalazine, ras-selective lethal small molecule 3 (RSL-3), and cyst(e)inases have an obvious effect on radiation sensitization; such sensitization is mainly related to ROS accumulation and lipid peroxidation [18].
The radiation resistance of hypoxic tumor cells is closely related to antioxidant stimulation. For example, hypoxia-inducible factor 1 (HIF-1) enhances the activities of glycolysis, serine synthesis, and pentose phosphate pathways. In addition, it increases the production of antioxidants, thus buffering radiation-induced ROS and resulting in radiation resistance. In addition, hypoxia itself increases the production of ROS, triggering a feedback loop and stimulating a metabolic process conducive for generating antioxidants; it can activate autophagy to accelerate the removal of ROS products, thus making cells radiation-resistant [3][20]. However, due to the large accumulation of ROS, hypoxic tumor cells rely heavily on the antioxidant system to maintain homeostasis.
2. Hypoxia Protects Tumor Cells from Ferroptosis by Regulating Iron Homeostasis
Hypoxia has been recognized as one of the basic and important characteristics of solid tumors; it plays an important role in various physiological events, such as cell proliferation, angiogenesis, metabolism, tumor invasion, and metastasis [21]. HIF-1 is a key regulator of hypoxia response in tumor cells and plays a key role in the adaptation of tumor cells to the hypoxic microenvironment. Hypoxia and HIF-1 overexpression are associated with radiation and chemotherapy resistance, as well as poor clinical prognosis of solid tumors [21][22]. Radiation resistance caused by hypoxia is considered to be the main reason why radiation therapy for solid tumors fails to achieve the expected results. Different mechanisms have been proposed to explain radiation resistance caused by hypoxia, among which the oxygen fixation hypothesis is the most accepted [23]. In addition, the upregulation of HIF-1α and enhancement of antioxidant system activity caused by hypoxia are also considered to be important factors for radiation resistance [3][20]. Interestingly, recent studies have shown that hypoxia can protect tumor cells from ferroptosis [24], which may explain the poor prognosis of hypoxic tumors.
In the body, cellular iron is essential for maintaining various metabolic pathways. Excessive free iron (Fe3+ or Fe2+) may lead to oxidative damage and cell death [25]. In addition, intracellular free iron can induce ferroptosis by activating arachidonate lipoxygenases (ALOXs) and promoting ROS production [4][26]. Therefore, the maintenance of iron homeostasis plays a crucial role in cell development; ferritin plays the most important role in regulating intracellular iron homeostasis. Ferritin heavy chain (FTH), ferritin light chain (FTL), and mitochondrial ferritin (FTMT) are three known ferritin proteins that have ferroxidase activity which can store Fe3+ and reduce intracellular free Fe2+ levels [27], thus playing an important role in ferroptosis. The release of iron in ferritin is regulated by the process of ferritinophagy; nuclear receptor coactivator 4 (NCOA4), its main regulator, directly binds to FTH and transports the complex to the lysosome for degradation [28]. The results of Fuhrmann et al. demonstrated that under hypoxic conditions, the transcription rate of NCOA4 decreases, leading to an increased FTH and FTMT expression, which protects cells from ferroptosis [24] (Figure 1). Subsequently, this was verified in diabetic mice, osteoporotic mice, and other animal models [29][30]. Furthermore, it was proven that the downregulation of NCOA4 and inhibition of ferroptosis under hypoxic conditions were associated with poor prognosis of stomach adenocarcinoma and hepatocellular carcinoma [31][32]. In conclusion, hypoxia represses ferritinophagy-mediated ferroptosis and leads to unfavorable prognosis; correspondingly, the induction of ferritinophagy and disruption of iron homeostasis may be an effective way to improve the therapeutic effect of hypoxic tumor cells.
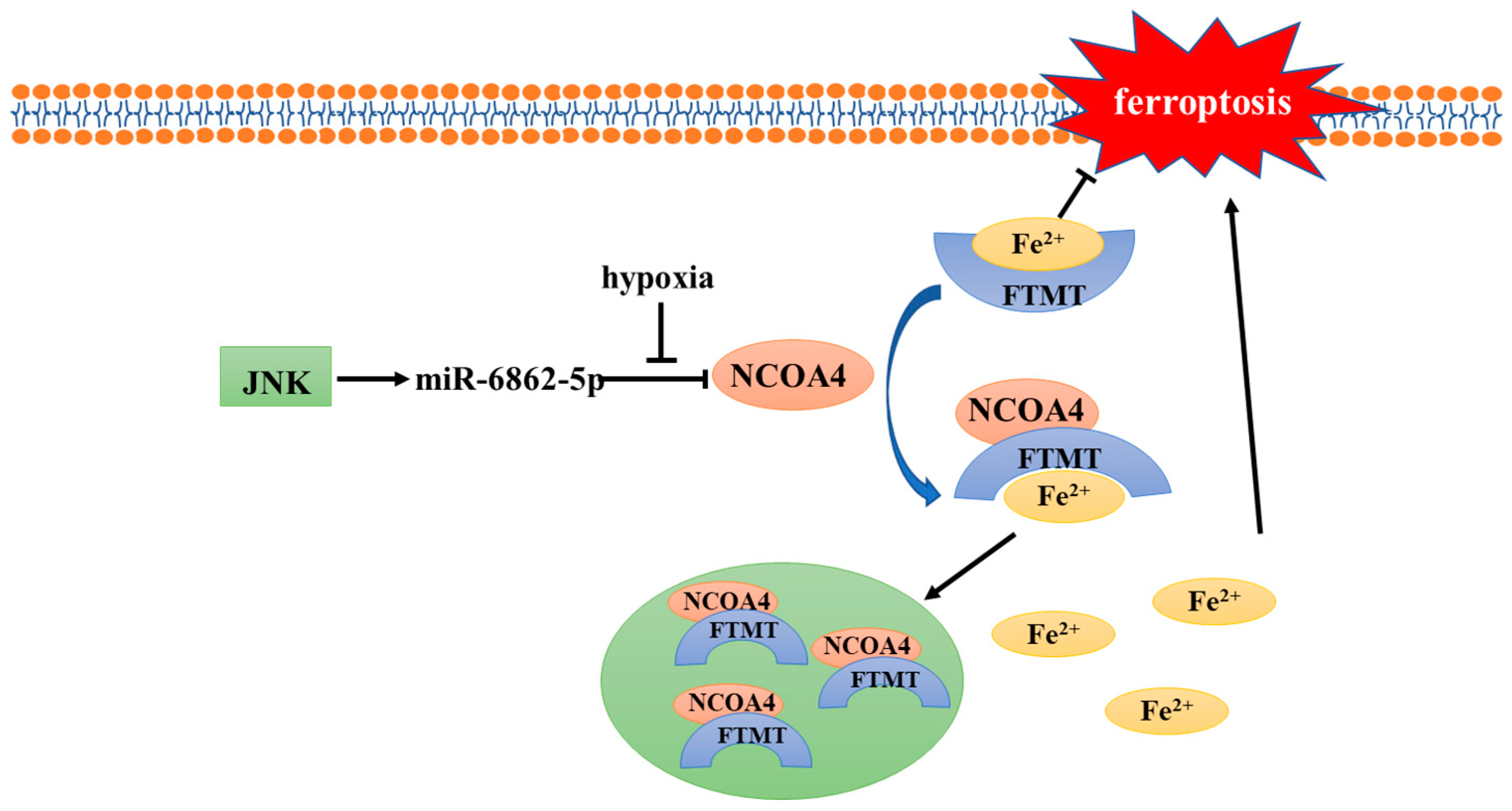
Figure 1. Mechanism of NCOA4 regulation under hypoxia.
The transcription of NCOA4 is related to miR-6862-5p. Under normoxic conditions, NCOA4 can bind with FTMT and Fe2+, and then send the complex to the autolysosome for degradation. However, the transcription of NCOA4 is inhibited when hypoxia occurs, thus blocking ferritinophagy and inhibiting ferroptosis. JNK, c-jun N-terminal kinase; NCOA4, nuclear receptor coactivator 4; FTMT, mitochondrial ferritin.
3. Bidirectional Regulation of Hypoxia-Inducible Factors on Ferroptosis
Hypoxia-inducible factors (HIFs) are ubiquitous in human and mammalian cells and can be stably expressed under hypoxic conditions. HIFs are heterodimeric transcription factors, composed of α and β subunits. The activity of HIFs is mainly dependent on α subunits, while β subunits are responsible for maintaining its stability [33]. The reason why α and β subunits function differently is that α subunits have an oxygen-dependent degradation domain (ODDD) in their structures [34]. In addition to ODDD, the α subunits have two transactivation domains: NH2-terminal (N-TAD) and COOH-terminal (C-TAD). They are responsible for the transcriptional activity of HIF-1/2α [35]. There are three subtypes of HIF-α: HIF-1α, HIF-2α, and HIF-3α [36][37]. HIF-1α and HIF-2α are particularly important for hypoxia response and can form complexes with HIF-1β [38]. The role of HIF-3α is unclear, but it has been suggested that HIF-3α may be involved in the transcriptional inhibition of HIF-1α [39].
The regulatory pathways of HIF-1/2α can be divided into oxygen-dependent and oxygen-independent pathways. Under normoxic conditions, proline residues (P402 and P564) within the ODDD of HIF-1α subunits and (P405 and P531) of HIF-2α subunits are hydroxylated by prolyl hydroxylases domain enzymes (PHD1-4) [40][41]. Hydroxylated HIF-1/2α can be recognized and ubiquitinated by the von Hippel–Lindau protein (pVHL), which mediates the assembly of the VHL/elongin C/elongin B/cullin-2 E3 ubiquitin ligase complex and is subsequently degraded by proteasomes [42]. In addition to prolyl hydroxylase, factor inhibiting HIF (FIH) is another cellular dioxygenase which could hydroxylate the 803 and 847 residues of asparagine within the C-TADs of HIF-1α and HIF-2α, respectively, and inhibit the transcription of HIF-1/2α [43]. However, PHDs and FIH are both oxygen dependent. Under hypoxic conditions, their activity is inhibited, allowing HIF-1/2α to escape ubiquitination and be transported to the nucleus after binding with HIF-1β [44][45]. In addition, ROS can also regulate the activity of PHDs and FIH. Studies have shown that NADPH oxidase 1 (NOX1) and NADPH oxidase 4 (NOX4) maintain HIF-2α expression in renal carcinoma through ROS generation, contributing to renal carcinogenesis [46]. Moreover, in recent years, several studies have shown that HIF can be regulated without oxygen; for example, heat shock protein 90 (Hsp90) can bind to HIF-1/2α subunits to inhibit their degradation [47], PI3K/AKT and MAPK/ERK pathways participate in the regulation of HIF-1/2α expression [48][49], and post-transcriptional acetylation and deacetylation regulate HIF-1/2α transcription and stability [50][51].
HIF-1/2α play a crucial role in the response of tumors to low oxygen [52]. Furthermore, the expression of HIF-1/2α is often associated with poor prognosis and chemoradiotherapy resistance [53]. Therefore, inhibiting HIF-1/2α expression and their downstream pathways activation is considered to be a reliable way to enhance radiosensitivity. Recently, HIF-1/2α subunits were found to play an important role in the regulation of ferroptosis (Figure 2), which further proved that ferroptosis inhibition is an important factor leading to unfavorable prognosis of hypoxic tumors.
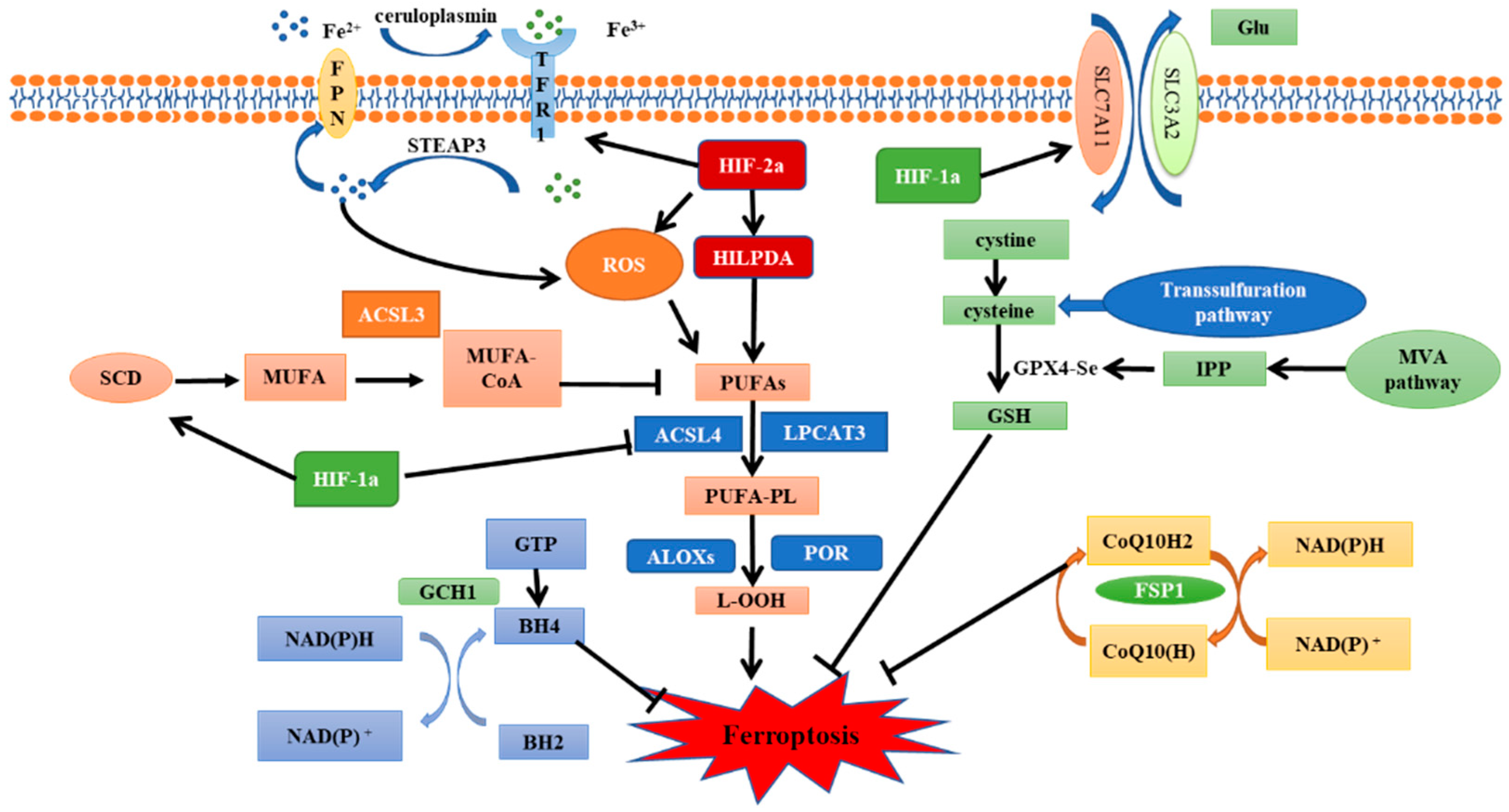
Figure 2. The regulatory pathways of ferroptosis and the role of HIF.
Ferroptosis is regulated by iron metabolism, lipid metabolism and antioxidant system. SLC7A11-GPX4-GSH pathway, FSP1-CoQ10-NAD (P)H pathway and GCH1-BH4-DHFR pathway are several classic ferroptosis inhibition pathways. HIF-1α has a significant inhibitory effect on ferroptosis. It can promote the expression of SCD1 to increase the synthesis of MUFA, inhibit the expression of ACSL4 to avoid lipid peroxidation, and reduce the degradation of SLC7A11 to enhance the antioxidant capacity of cells. In contrast, HIF-2α promotes ferroptosis. Activation of the HIF-2α-HILPDA pathway can promote the synthesis of PUFAs to induce lipid peroxidation. HIF-2α can also regulate iron metabolism gene to increase intracellular free iron content and oxidize cysteine to increase ROS production. SLC7A11, Cystine/glutamate antiporter solute carrier family 7 member 11; GPX4, glutathione peroxidase 4; GSH, glutathione; FSP1, ferroptosis suppressor protein 1; GGH1, GTP Cyclohydrolase 1; BH4, tetrahydrobiopterin; DHFR, dihydrofolate reductase; HIF, hypoxia-inducible factor; SCD1, stearoyl-CoA desaturase 1; MUFA, monounsaturated fatty acids; ACSL4, acyl-CoA synthetase long-chain family member 4; HILPDA, hypoxia inducible lipid droplet-associated proteins; and PUFAs, polyunsaturated fatty acids.
3.1. HIF-1α—A Negative Regulator of Ferroptosis
HIF-1α plays an important role in the metabolic regulation of hypoxic cells. Activation of HIF-1α can enhance the activities of glycolysis, serine synthesis, and pentose phosphate pathways [54], thereby allowing cells to adapt to the hypoxic environment. The massive production of antioxidants induced by HIF-1α is also considered as one of the mechanisms of radiation resistance in hypoxic cells. In recent years, it has been found that HIF-1α expression is associated with ferroptosis.
To begin with, HIF-1α can inhibit ferroptosis by regulating SLC7A11. SLC7A11 expression is positively associated with HIF-1α in human hepatocellular carcinoma (HCC) tissues [55]. It has been shown that HIF-1α can inhibit methyltransferase-like 14 (METTL14) expression, thus inhibiting the degradation of SLC7A11 mRNA and reducing the sensitivity of HCC cells to ferroptosis [56]. Moreover, the HIF-1α/SLC7A11 pathway has also been reported in the nervous system and oral squamous cell carcinoma (OSCC) [57][58][59]. Unfortunately, one study found that HIF-1α does not regulate SLC7A11 in breast cancer cells [60]. Therefore, the application scope and specific mechanism of this pathway need to be further studied. HIF-1α can also inhibit ferroptosis by regulating lipid metabolism. Yang et al. demonstrated in a variety of human tumor cell lines that aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL, also call BMAL1) inhibits ferroptosis by inhibiting EGLN2 (also call PHD2) activation and activating HIF-1α. Additionally, HIF-1α-induced ferroptosis inhibition may be related to its role in promoting lipid storage [61]. Conversely, EGLN1 (also call PHD1) inhibits ferroptosis by promoting lymphoid-specific helicase (LSH) expression in lung cancer cell lines [62]. This result further proves that the regulation of HIF-1α on ferroptosis is complex and variable. Furthermore, ACSL4 is a key enzyme in the formation of polyunsaturated fatty acids; HIF-1α inhibits the synthesis of ACSL4 to inhibit ferroptosis [63]. In nasopharyngeal carcinoma (NPC), HIF-1α upregulates the expression of stearoyl-CoA desaturase 1 (SCD1), which mediates the production of monounsaturated fatty acids and inhibits ferroptosis [64]. Finally, a study of cervical cancer (CC) demonstrated that HIF-1α activation can suppress ferroptosis by destroying iron homeostasis imbalance [65].
These findings confirm that HIF-1α plays an important role in the regulation of ferroptosis. Under hypoxic conditions, HIF-1α can produce antioxidants to resist oxidative stress caused by radiation-induced ROS. It has been confirmed that IR can also cause significant upregulation of HIF-1α, thus further aggravating radiation resistance of hypoxic cells [66][67]. Therefore, HIF-1α is an effective target for radiation sensitization of hypoxic cells [68][69]. Ferroptosis accounts for a large proportion of IR-induced cell death, even more than apoptosis and necrosis [19]. HIF-1α-mediated ferroptosis inhibition is closely associated with poor prognosis and treatment tolerance of hypoxic tumors [59]. Consequently, it may be practical to reduce HIF-1α-induced radiation resistance by regulating the occurrence of ferroptosis.
3.2. HIF-2α—A Positive Regulator of Ferroptosis
As two important members of the HIF family, both HIF-1α and HIF-2α share 48% structural homology [70]. Thus, they have certain functional similarities in regulating physiologic and pathologic responses in hypoxic environments and in affecting the development of hypoxia-related diseases. However, increasing evidence has revealed that when expressed in the same cell type, HIF-1α and HIF-2α activation can produce highly different or even opposite results [71][72]. Previous studies on ferroptosis have shown that HIF-2α plays a positive role in ferroptosis in various tumor models [73][74].
Zou et al. showed that HIF-2α activation stimulated hypoxia-induced expression of lipid droplet-associated proteins (HILPDA) and selectively enriched polyunsaturated lipids to improve the sensitivity of clear-cell carcinoma cells to ferroptosis. Notably, HIF-1α also plays a role in increasing ferroptosis sensitivity in ovarian clear cell carcinoma cells [73]. The clear-cell carcinomas (CCCs) are always associated with highly active lipid and glycogen synthesis and deposition, which promotes tumor progression and treatment resistance [75]. HIF-mediated ferroptosis susceptibility may be related to the special metabolic state and high GPX4 dependence of clear-cell carcinoma cells [76]. Therefore, the relationship between HIF and ferroptosis in other tumors with high metabolic activities and GPX4 dependence is worth exploring. In addition, under hypoxic conditions, HIF-2α is a major regulator of erythropoiesis and cellular iron metabolism, which can interfere with bone morphogenetic protein (BMP) signaling, inhibit the expression of hepcidin, and regulate iron uptake and mobilization in the intestinal diet [77][78]. Studies have shown that HIF-2α activation can upregulate the expression of lipid and iron regulation genes as well as increase the intracellular iron level, thereby leading to the susceptibility of colorectal cancer cells to ferroptosis. Furthermore, HIF-2α activation enhances colon cancer cell death by promoting irreversible cysteine oxidation in an iron-dependent manner to increase ROS production. In contrast, HIF-1α has no significant regulatory effect on colorectal cancers [74]. A recent study showed that HIF-2α also mediates ferroptosis in chondrocytes [79], implying a more general role of HIF-2α in ferroptosis in other contexts. However, as a hypoxia-triggered factor, HIF-2α has a similar effect as HIF-1α on radiation resistance, so the role of HIF-2α-mediated ferroptosis in radiotherapy is open to debate.
Radiation and chemotherapy resistance caused by hypoxia has always been a major problem in tumor therapy; however, targeting hypoxic cells remains a challenge, as frequent use of HIF inhibitors can rapidly develop drug resistance in tumor cells. Recent studies have shown that regulating the activity of HIF to promote ferroptosis sensitivity may be a good way to solve the poor prognosis of hypoxic tumors [59]. As mentioned above, HIF-1α inhibits ferroptosis by targeting SLC7A11, lipid metabolism, and iron homeostasis. HIF-2α induces ferroptosis, perhaps because it is a major regulator of iron metabolism in hypoxic conditions. However, both HIF-1α and HIF-2α show promoting effects on ferroptosis in CCCs, suggesting the regulation of ferroptosis by HIF is complex and can be affected by many factors such as tumor types, metabolic levels and external environment. Therefore, further studies on HIF and ferroptosis should be carried out to provide more effective methods for the radiosensitization of solid tumors.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment. Cancer 2005, 104, 1129–1137.
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437.
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88.
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285.
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609.
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98.
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309.
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194.
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396.
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331.
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698.
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590.
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523.
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53.
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685.
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162.
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112.
- Yang, G.; Shi, R.; Zhang, Q. Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression. Int. J. Mol. Sci. 2020, 21, 8162.
- Ruan, K.; Song, G.; Ouyang, G. Role of hypoxia in the hallmarks of human cancer. J. Cell. Biochem. 2009, 107, 1053–1062.
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447.
- Fuhrmann, D.C.; Mondorf, A.; Beifuß, J.; Jung, M.; Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020, 36, 101670.
- Zhou, Y.; Que, K.T.; Zhang, Z.; Yi, Z.J.; Zhao, P.X.; You, Y.; Gong, J.P.; Liu, Z.J. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. 2018, 7, 4012–4022.
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e626.
- Hilton, R.J.; David Andros, N.; Watt, R.K. The ferroxidase center is essential for ferritin iron loading in the presence of phosphate and minimizes side reactions that form Fe(III)-phosphate colloids. Biometals 2012, 25, 259–273.
- Gryzik, M.; Srivastava, A.; Longhi, G.; Bertuzzi, M.; Gianoncelli, A.; Carmona, F.; Poli, M.; Arosio, P. Expression and characterization of the ferritin binding domain of Nuclear Receptor Coactivator-4 (NCOA4). Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2710–2716.
- Ni, S.; Yuan, Y.; Qian, Z.; Zhong, Z.; Lv, T.; Kuang, Y.; Yu, B. Hypoxia inhibits RANKL-induced ferritinophagy and protects osteoclasts from ferroptosis. Free Radic. Biol. Med. 2021, 169, 271–282.
- Li, W.; Li, W.; Wang, Y.; Leng, Y.; Xia, Z. Inhibition of DNMT-1 alleviates ferroptosis through NCOA4 mediated ferritinophagy during diabetes myocardial ischemia/reperfusion injury. Cell Death Discov. 2021, 7, 267.
- Xiao, R.; Wang, S.; Guo, J.; Liu, S.; Ding, A.; Wang, G.; Li, W.; Zhang, Y.; Bian, X.; Zhao, S.; et al. Ferroptosis-related gene NOX4, CHAC1 and HIF1A are valid biomarkers for stomach adenocarcinoma. J. Cell. Mol. Med. 2022, 26, 1183–1193.
- Wen, K.; Yan, Y.; Shi, J.; Hu, L.; Wang, W.; Liao, H.; Li, H.; Zhu, Y.; Mao, K.; Xiao, Z. Construction and Validation of a Combined Ferroptosis and Hypoxia Prognostic Signature for Hepatocellular Carcinoma. Front. Mol. Biosci. 2021, 8, 809672.
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514.
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340.
- Li, H.; Ko, H.P.; Whitlock, J.P. Induction of phosphoglycerate kinase 1 gene expression by hypoxia. Roles of Arnt and HIF1alpha. J. Biol. Chem. 1996, 271, 21262–21267.
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278.
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087.
- Gordan, J.D.; Simon, M.C. Hypoxia-inducible factors: Central regulators of the tumor phenotype. Curr. Opin. Genet. Dev. 2007, 17, 71–77.
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001, 414, 550–554.
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472.
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206.
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741.
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471.
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468.
- Chilov, D.; Camenisch, G.; Kvietikova, I.; Ziegler, U.; Gassmann, M.; Wenger, R.H. Induction and nuclear translocation of hypoxia-inducible factor-1 (HIF-1): Heterodimerization with ARNT is not necessary for nuclear accumulation of HIF-1alpha. J. Cell Sci. 1999, 112 Pt 8, 1203–1212.
- Block, K.; Gorin, Y.; Hoover, P.; Williams, P.; Chelmicki, T.; Clark, R.A.; Yoneda, T.; Abboud, H.E. NAD(P)H oxidases regulate HIF-2alpha protein expression. J. Biol. Chem. 2007, 282, 8019–8026.
- Katschinski, D.M.; Le, L.; Heinrich, D.; Wagner, K.F.; Hofer, T.; Schindler, S.G.; Wenger, R.H. Heat induction of the unphosphorylated form of hypoxia-inducible factor-1alpha is dependent on heat shock protein-90 activity. J. Biol. Chem. 2002, 277, 9262–9267.
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545.
- Chang, H.; Shyu, K.G.; Lin, S.; Tsai, S.C.; Wang, B.W.; Liu, Y.C.; Sung, Y.L.; Lee, C.C. The plasminogen activator inhibitor-1 gene is induced by cell adhesion through the MEK/ERK pathway. J. Biomed. Sci. 2003, 10, 738–745.
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720.
- Chen, R.; Xu, M.; Hogg, R.T.; Li, J.; Little, B.; Gerard, R.D.; Garcia, J.A. The acetylase/deacetylase couple CREB-binding protein/Sirtuin 1 controls hypoxia-inducible factor 2 signaling. J. Biol. Chem. 2012, 287, 30800–30811.
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162.
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682.
- de Heer, E.C.; Jalving, M.; Harris, A.L. HIFs, angiogenesis, and metabolism: Elusive enemies in breast cancer. J. Clin. Investig. 2020, 130, 5074–5087.
- He, Q.; Liu, M.; Huang, W.; Chen, X.; Zhang, B.; Zhang, T.; Wang, Y.; Liu, D.; Xie, M.; Ji, X.; et al. IL-1β-Induced Elevation of Solute Carrier Family 7 Member 11 Promotes Hepatocellular Carcinoma Metastasis Through Up-regulating Programmed Death Ligand 1 and Colony-Stimulating Factor 1. Hepatology 2021, 74, 3174–3193.
- Fan, Z.; Yang, G.; Zhang, W.; Liu, Q.; Liu, G.; Liu, P.; Xu, L.; Wang, J.; Yan, Z.; Han, H.; et al. Hypoxia blocks ferroptosis of hepatocellular carcinoma via suppression of METTL14 triggered YTHDF2-dependent silencing of SLC7A11. J. Cell. Mol. Med. 2021, 25, 10197–10212.
- Sims, B.; Clarke, M.; Francillion, L.; Kindred, E.; Hopkins, E.S.; Sontheimer, H. Hypoxic preconditioning involves system Xc- regulation in mouse neural stem cells. Stem Cell Res. 2012, 8, 285–291.
- Hsieh, C.H.; Lin, Y.J.; Chen, W.L.; Huang, Y.C.; Chang, C.W.; Cheng, F.C.; Liu, R.S.; Shyu, W.C. HIF-1α triggers long-lasting glutamate excitotoxicity via system x(c)(-) in cerebral ischaemia-reperfusion. J. Pathol. 2017, 241, 337–349.
- Yang, Y.; Tang, H.; Zheng, J.; Yang, K. The PER1/HIF-1alpha negative feedback loop promotes ferroptosis and inhibits tumor progression in oral squamous cell carcinoma. Transl. Oncol. 2022, 18, 101360.
- Briggs, K.J.; Koivunen, P.; Cao, S.; Backus, K.M.; Olenchock, B.A.; Patel, H.; Zhang, Q.; Signoretti, S.; Gerfen, G.J.; Richardson, A.L.; et al. Paracrine Induction of HIF by Glutamate in Breast Cancer: EglN1 Senses Cysteine. Cell 2016, 166, 126–139.
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 2019, 5, eaaw2238.
- Jiang, Y.; Mao, C.; Yang, R.; Yan, B.; Shi, Y.; Liu, X.; Lai, W.; Liu, Y.; Wang, X.; Xiao, D.; et al. EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics 2017, 7, 3293–3305.
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321.
- Yin, H.; Qiu, X.; Shan, Y.; You, B.; Xie, L.; Zhang, P.; Zhao, J.; You, Y. HIF-1α downregulation of miR-433-3p in adipocyte-derived exosomes contributes to NPC progression via targeting SCD1. Cancer Sci. 2021, 112, 1457–1470.
- Xiong, J.; Nie, M.; Fu, C.; Chai, X.; Zhang, Y.; He, L.; Sun, S. Hypoxia Enhances HIF1α Transcription Activity by Upregulating KDM4A and Mediating H3K9me3, Thus Inducing Ferroptosis Resistance in Cervical Cancer Cells. Stem Cells Int. 2022, 2022, 1608806.
- Kim, W.; Kim, M.S.; Kim, H.J.; Lee, E.; Jeong, J.H.; Park, I.; Jeong, Y.K.; Jang, W.I. Role of HIF-1α in response of tumors to a combination of hyperthermia and radiation in vivo. Int. J. Hyperth. 2018, 34, 276–283.
- Semenza, G.L. Intratumoral hypoxia, radiation resistance, and HIF-1. Cancer Cell 2004, 5, 405–406.
- Ikezawa, Y.; Sakakibara-Konishi, J.; Mizugaki, H.; Oizumi, S.; Nishimura, M. Inhibition of Notch and HIF enhances the antitumor effect of radiation in Notch expressing lung cancer. Int. J. Clin. Oncol. 2017, 22, 59–69.
- Moeller, B.J.; Dewhirst, M.W. HIF-1 and tumour radiosensitivity. Br. J. Cancer 2006, 95, 1–5.
- Zhao, J.; Du, F.; Shen, G.; Zheng, F.; Xu, B. The role of hypoxia-inducible factor-2 in digestive system cancers. Cell Death Dis. 2015, 6, e1600.
- Imamura, T.; Kikuchi, H.; Herraiz, M.T.; Park, D.Y.; Mizukami, Y.; Mino-Kenduson, M.; Lynch, M.P.; Rueda, B.R.; Benita, Y.; Xavier, R.J.; et al. HIF-1alpha and HIF-2alpha have divergent roles in colon cancer. Int. J. Cancer 2009, 124, 763–771.
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22.
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1617.
- Singhal, R.; Mitta, S.R.; Das, N.K.; Kerk, S.A.; Sajjakulnukit, P.; Solanki, S.; Andren, A.; Kumar, R.; Olive, K.P.; Banerjee, R.; et al. HIF-2α activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Investig. 2021, 131, e143691.
- Wettersten, H.I.; Aboud, O.A.; Lara, P.N., Jr.; Weiss, R.H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 410–419.
- Miess, H.; Dankworth, B.; Gouw, A.M.; Rosenfeldt, M.; Schmitz, W.; Jiang, M.; Saunders, B.; Howell, M.; Downward, J.; Felsher, D.W.; et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 2018, 37, 5435–5450.
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J. Clin. Investig. 2009, 119, 1159–1166.
- Duarte, T.L.; Talbot, N.P.; Drakesmith, H. NRF2 and Hypoxia-Inducible Factors: Key Players in the Redox Control of Systemic Iron Homeostasis. Antioxid. Redox Signal. 2021, 35, 433–452.
- Zhou, X.; Zheng, Y.; Sun, W.; Zhang, Z.; Liu, J.; Yang, W.; Yuan, W.; Yi, Y.; Wang, J.; Liu, J. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2α-dependent manner. Cell Prolif. 2021, 54, e13134.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
926
Revisions:
2 times
(View History)
Update Date:
16 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No