 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Farhoud Faraji | -- | 3161 | 2022-05-10 16:02:14 | | | |
| 2 | Farhoud Faraji | Meta information modification | 3161 | 2022-05-10 17:13:18 | | | | |
| 3 | Beatrix Zheng | -6 word(s) | 3155 | 2022-05-11 03:43:56 | | |
Video Upload Options
The Hippo signaling pathway, originally discovered as a mechanism regulating tissue growth and organ size, transduces intracellular and extracellular signals to regulate the transcriptional co-activators YAP and TAZ. Alterations in the Hippo pathway resulting in persistent YAP and TAZ activation have emerged as major oncogenic drivers. The researchers' analysis of the human Head and neck squamous cell carcinoma (HNSCC) oncogenome revealed multiple genomic alterations impairing Hippo signaling and activating YAP and TAZ, which in turn contribute to HNSCC development. This includes mutations and deletions of the FAT1 gene (29%) and amplification of the WWTR1 (encoding TAZ, 14%) and YAP1 genes (8%), together representing one of the most genetically altered signaling mechanisms in this malignancy.
1. Mechanisms of YAP/TAZ Activation in Head and Neck Squamous Cell Carcinoma (HNSCC)

1.1. FAT1: A Membrane-Associated Proto-Cadherin Assembling the Hippo Signalome
1.2. FAT1-Independent Mechanisms of Oncogenic Hippo Pathway Perturbation in HNSCC
2. Consequences YAP/TAZ Activation in HNSCC
2.1. Cancer Stemness
2.2. Tumor Progression and Poor Prognosis
2.3. Therapeutic Resistance
3. YAP/TAZ Activation Exposes HNSCC Vulnerabilities and Therapeutic Opportunities
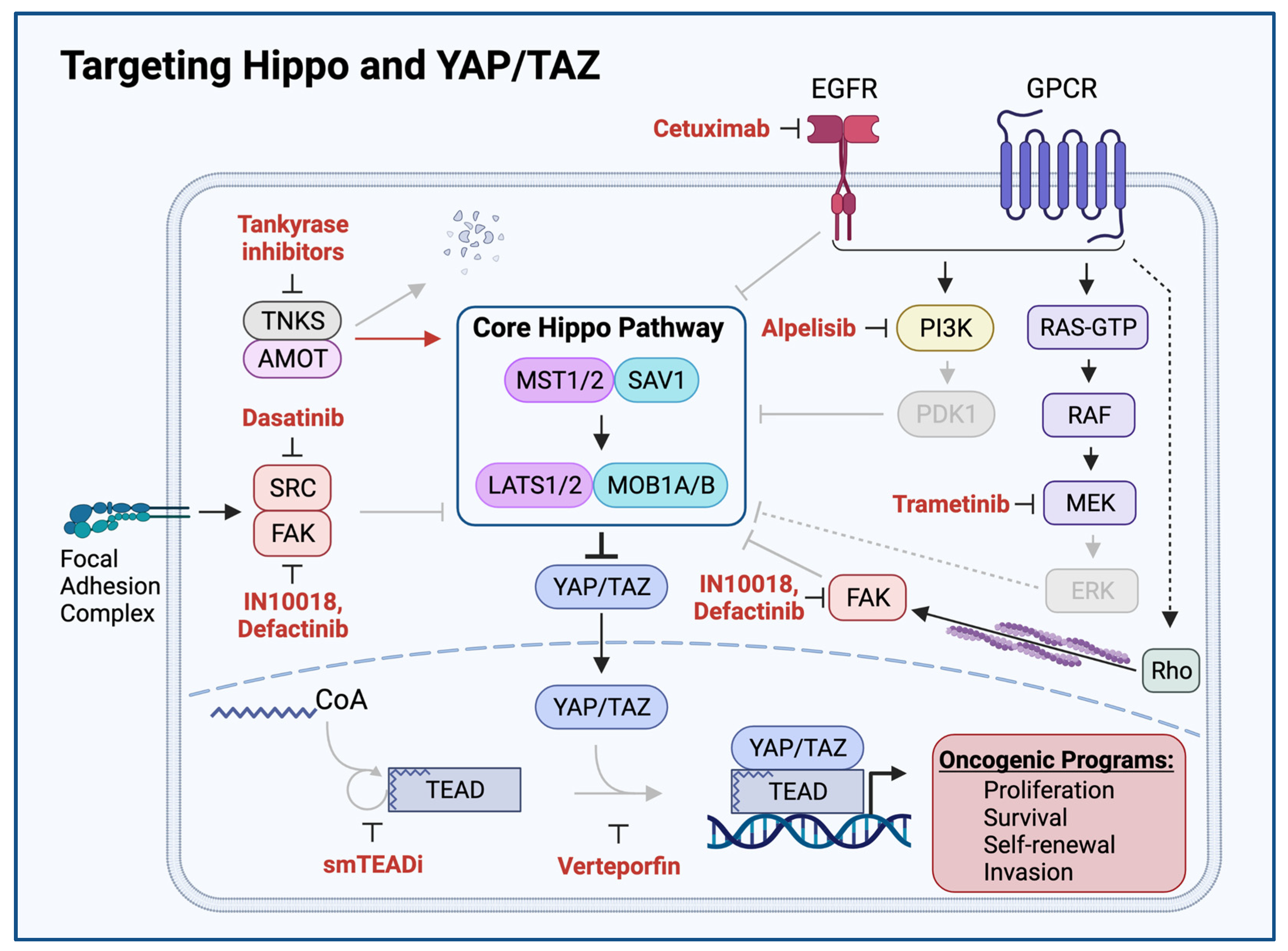
References
- Martin, D.; Degese, M.S.; Vitale-Cross, L.; Iglesias-Bartolome, R.; Valera, J.L.C.; Wang, Z.; Feng, X.; Yeerna, H.; Vadmal, V.; Moroishi, T.; et al. Assembly and activation of the Hippo signalome by FAT1 tumor suppressor. Nat. Commun. 2018, 9, 2372.
- Morris, L.G.T.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.A.; Turcan, Ş.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261.
- Pastushenko, I.; Mauri, F.; Song, Y.; de Cock, F.; Meeusen, B.; Swedlund, B.; Impens, F.; Van Haver, D.; Opitz, M.; Thery, M.; et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 2021, 589, 448–455.
- Wang, Z.; Wu, V.H.; Allevato, M.M.; Gilardi, M.; He, Y.; Luis Callejas-Valera, J.; Vitale-Cross, L.; Martin, D.; Amornphimoltham, P.; Mcdermott, J.; et al. Syngeneic animal models of tobacco-associated oral cancer reveal the activity of in situ anti-CTLA-4. Nat. Commun. 2019, 10, 5546.
- Mann, J.E.; Kulkarni, A.; Birkeland, A.C.; Kafelghazal, J.; Eisenberg, J.; Jewell, B.M.; Ludwig, M.L.; Spector, M.E.; Jiang, H.; Carey, T.E.; et al. The molecular landscape of the University of Michigan laryngeal squamous cell carcinoma cell line panel. Head Neck 2019, 41, 3114–3124.
- Katoh, M. Function and cancer genomics of FAT family genes (review). Int. J. Oncol. 2012, 41, 1913–1918.
- Kim, K.T.; Kim, B.-S.; Kim, J.H. Association between FAT1 mutation and overall survival in patients with human papillomavirus-negative head and neck squamous cell carcinoma. Head Neck 2016, 38 (Suppl. 1), E2021–E2029.
- Kriegs, M.; Clauditz, T.S.; Hoffer, K.; Bartels, J.; Buhs, S.; Gerull, H.; Zech, H.B.; Bußmann, L.; Struve, N.; Rieckmann, T.; et al. Analyzing expression and phosphorylation of the EGF receptor in HNSCC. Sci. Rep. 2019, 9, 13564.
- Temam, S.; Kawaguchi, H.; El-Naggar, A.K.; Jelinek, J.; Tang, H.; Liu, D.D.; Lang, W.; Issa, J.-P.; Lee, J.J.; Mao, L. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 2164–2170.
- Fan, R.; Kim, N.-G.; Gumbiner, B.M. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl. Acad. Sci. USA 2013, 110, 2569–2574.
- Ando, T.; Arang, N.; Wang, Z.; Costea, D.E.; Feng, X.; Goto, Y.; Izumi, H.; Gilardi, M.; Ando, K.; Gutkind, J.S. EGFR Regulates the Hippo pathway by promoting the tyrosine phosphorylation of MOB1. Commun. Biol. 2021, 4, 1237.
- Omori, H.; Nishio, M.; Masuda, M.; Miyachi, Y.; Ueda, F.; Nakano, T.; Sato, K.; Mimori, K.; Taguchi, K.; Hikasa, H.; et al. YAP1 is a potent driver of the onset and progression of oral squamous cell carcinoma. Sci. Adv. 2020, 6, eaay3324.
- Bichsel, S.J.; Tamaskovic, R.; Stegert, M.R.; Hemmings, B.A. Mechanism of activation of NDR (nuclear Dbf2-related) protein kinase by the hMOB1 protein. J. Biol. Chem. 2004, 279, 35228–35235.
- Zhang, L.; Tang, F.; Terracciano, L.; Hynx, D.; Kohler, R.; Bichet, S.; Hess, D.; Cron, P.; Hemmings, B.A.; Hergovich, A.; et al. NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr. Biol. CB 2015, 25, 296–305.
- Enomoto, A.; Kido, N.; Ito, M.; Morita, A.; Matsumoto, Y.; Takamatsu, N.; Hosoi, Y.; Miyagawa, K. Negative regulation of MEKK1/2 signaling by serine-threonine kinase 38 (STK38). Oncogene 2008, 27, 1930–1938.
- Hergovich, A. The Roles of NDR Protein Kinases in Hippo Signalling. Genes 2016, 7, 21.
- Cheng, H.; Yang, X.; Si, H.; Saleh, A.D.; Xiao, W.; Coupar, J.; Gollin, S.M.; Ferris, R.L.; Issaeva, N.; Yarbrough, W.G.; et al. Genomic and Transcriptomic Characterization Links Cell Lines with Aggressive Head and Neck Cancers. Cell Rep. 2018, 25, 1332–1345.e5.
- Li, J.; Li, Z.; Wu, Y.; Wang, Y.; Wang, D.; Zhang, W.; Yuan, H.; Ye, J.; Song, X.; Yang, J.; et al. The Hippo effector TAZ promotes cancer stemness by transcriptional activation of SOX2 in head neck squamous cell carcinoma. Cell Death Dis. 2019, 10, 603.
- Li, Z.; Wang, Y.; Zhu, Y.; Yuan, C.; Wang, D.; Zhang, W.; Qi, B.; Qiu, J.; Song, X.; Ye, J.; et al. The Hippo transducer TAZ promotes epithelial to mesenchymal transition and cancer stem cell maintenance in oral cancer. Mol. Oncol. 2015, 9, 1091–1105.
- Truong, A.B.; Kretz, M.; Ridky, T.W.; Kimmel, R.; Khavari, P.A. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006, 20, 3185–3197.
- Euskirchen, G.; Auerbach, R.K.; Snyder, M. SWI/SNF chromatin-remodeling factors: Multiscale analyses and diverse functions. J. Biol. Chem. 2012, 287, 30897–30905.
- Saladi, S.V.; Ross, K.; Karaayvaz, M.; Tata, P.R.; Mou, H.; Rajagopal, J.; Ramaswamy, S.; Ellisen, L.W. ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 2017, 31, 35–49.
- Moleirinho, S.; Chang, N.; Sims, A.H.; Tilston-Lünel, A.M.; Angus, L.; Steele, A.; Boswell, V.; Barnett, S.C.; Ormandy, C.; Faratian, D.; et al. KIBRA exhibits MST-independent functional regulation of the Hippo signaling pathway in mammals. Oncogene 2013, 32, 1821–1830.
- Yu, J.; Zheng, Y.; Dong, J.; Klusza, S.; Deng, W.-M.; Pan, D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev. Cell 2010, 18, 288–299.
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803.
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419.
- Hiemer, S.E.; Zhang, L.; Kartha, V.K.; Packer, T.S.; Almershed, M.; Noonan, V.; Kukuruzinska, M.; Bais, M.V.; Monti, S.; Varelas, X. A YAP/TAZ-Regulated Molecular Signature Is Associated with Oral Squamous Cell Carcinoma. Mol. Cancer Res. MCR 2015, 13, 957–968.
- Wang, Y.; Gersten, A.; Moleirinho, S.; Gunn-Moore, F.J.; Reynolds, P.A.; Prystowsky, M.B. Fibroblasts in Head and Neck Squamous Cell Carcinoma Associated With Perineural Invasion Have High-Level Nuclear Yes-Associated Protein (YAP) Expression. Acad. Pathol. 2015, 2, 2374289515616972.
- Ge, L.; Smail, M.; Meng, W.; Shyr, Y.; Ye, F.; Fan, K.-H.; Li, X.; Zhou, H.-M.; Bhowmick, N.A. Yes-associated protein expression in head and neck squamous cell carcinoma nodal metastasis. PLoS ONE 2011, 6, e27529.
- Wei, Z.; Wang, Y.; Li, Z.; Yuan, C.; Zhang, W.; Wang, D.; Ye, J.; Jiang, H.; Wu, Y.; Cheng, J. Overexpression of Hippo pathway effector TAZ in tongue squamous cell carcinoma: Correlation with clinicopathological features and patients’ prognosis. J. Oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2013, 42, 747–754.
- Eun, Y.-G.; Lee, D.; Lee, Y.C.; Sohn, B.H.; Kim, E.H.; Yim, S.Y.; Kwon, K.H.; Lee, J.-S. Clinical significance of YAP1 activation in head and neck squamous cell carcinoma. Oncotarget 2017, 8, 111130–111143.
- García-Escudero, R.; Segrelles, C.; Dueñas, M.; Pombo, M.; Ballestín, C.; Alonso-Riaño, M.; Nenclares, P.; Álvarez-Rodríguez, R.; Sánchez-Aniceto, G.; Ruíz-Alonso, A.; et al. Overexpression of PIK3CA in head and neck squamous cell carcinoma is associated with poor outcome and activation of the YAP pathway. Oral Oncol. 2018, 79, 55–63.
- Pfister, D.G.; Spencer, S.; Adelstein, D.; Adkins, D.; Anzai, Y.; Brizel, D.M.; Bruce, J.Y.; Busse, P.M.; Caudell, J.J.; Cmelak, A.J.; et al. Head and Neck Cancers, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 873–898.
- Yoshikawa, K.; Noguchi, K.; Nakano, Y.; Yamamura, M.; Takaoka, K.; Hashimoto-Tamaoki, T.; Kishimoto, H. The Hippo pathway transcriptional co-activator, YAP, confers resistance to cisplatin in human oral squamous cell carcinoma. Int. J. Oncol. 2015, 46, 2364–2370.
- Shriwas, O.; Arya, R.; Mohanty, S.; Mohapatra, P.; Kumar, S.; Rath, R.; Kaushik, S.R.; Pahwa, F.; Murmu, K.C.; Majumdar, S.K.D.; et al. RRBP1 rewires cisplatin resistance in oral squamous cell carcinoma by regulating Hippo pathway. Br. J. Cancer 2021, 124, 2004–2016.
- Ehsanian, R.; Brown, M.; Lu, H.; Yang, X.P.; Pattatheyil, A.; Yan, B.; Duggal, P.; Chuang, R.; Doondeea, J.; Feller, S.; et al. YAP dysregulation by phosphorylation or ΔNp63-mediated gene repression promotes proliferation, survival and migration in head and neck cancer subsets. Oncogene 2010, 29, 6160–6171.
- Albanell, J.; Codony-Servat, J.; Rojo, F.; Del Campo, J.M.; Sauleda, S.; Anido, J.; Raspall, G.; Giralt, J.; Roselló, J.; Nicholson, R.I.; et al. Activated extracellular signal-regulated kinases: Association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor treatments. Cancer Res. 2001, 61, 6500–6510.
- Søland, T.M.; Husvik, C.; Koppang, H.S.; Boysen, M.; Sandvik, L.; Clausen, O.P.F.; Christoffersen, T.; Bryne, M. A study of phosphorylated ERK1/2 and COX-2 in early stage (T1-T2) oral squamous cell carcinomas. J. Oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2008, 37, 535–542.
- Judd, N.P.; Winkler, A.E.; Murillo-Sauca, O.; Brotman, J.J.; Law, J.H.; Lewis, J.S.; Dunn, G.P.; Bui, J.D.; Sunwoo, J.B.; Uppaluri, R. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res. 2012, 72, 365–374.
- Uppaluri, R.; Winkler, A.E.; Lin, T.; Law, J.H.; Haughey, B.H.; Nussenbaum, B.; Paniello, R.C.; Rich, J.T.; Diaz, J.A.; Michel, L.P.; et al. Biomarker and Tumor Responses of Oral Cavity Squamous Cell Carcinoma to Trametinib: A Phase II Neoadjuvant Window-of-Opportunity Clinical Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2186–2194.
- Zhao, Y.; Adjei, A.A. The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol. 2014, 11, 385–400.
- Mudianto, T.; Campbell, K.M.; Webb, J.; Zolkind, P.; Skidmore, Z.L.; Riley, R.; Barnell, E.K.; Ozgenc, I.; Giri, T.; Dunn, G.P.; et al. Yap1 Mediates Trametinib Resistance in Head and Neck Squamous Cell Carcinomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2326–2339.
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307.
- Zanconato, F.; Battilana, G.; Cordenonsi, M.; Piccolo, S. YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 2016, 29, 26–33.
- Bushweller, J.H. Targeting Transcription Factors in Cancer - from Undruggable to Reality. Nat Rev Cancer 2019, 19, 611–624.
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and Pharmacological Disruption of the TEAD-YAP Complex Suppresses the Oncogenic Activity of YAP. Genes Dev 2012, 26, 1300–1305.
- Schmidt-Erfurth, U.; Hasan, T. Mechanisms of Action of Photodynamic Therapy with Verteporfin for the Treatment of Age-Related Macular Degeneration. Surv Ophthalmol 2000, 45, 195–214.
- Gibault, F.; Corvaisier, M.; Bailly, F.; Huet, G.; Melnyk, P.; Cotelle, P. Non-Photoinduced Biological Properties of Verteporfin. Curr Med Chem 2016, 23, 1171–1184.
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet Lond. Engl. 2019, 394, 1915–1928.
- Tang, T.T.; Konradi, A.W.; Feng, Y.; Peng, X.; Ma, M.; Li, J.; Yu, F.-X.; Guan, K.-L.; Post, L. Small Molecule Inhibitors of TEAD Auto-palmitoylation Selectively Inhibit Proliferation and Tumor Growth of NF2-deficient Mesothelioma. Mol. Cancer Ther. 2021, 20, 986–998.
- Wang, W.; Li, N.; Li, X.; Tran, M.K.; Han, X.; Chen, J. Tankyrase Inhibitors Target YAP by Stabilizing Angiomotin Family Proteins. Cell Rep. 2015, 13, 524–532.
- Araujo, J.; Logothetis, C. Dasatinib: A potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat. Rev. 2010, 36, 492–500.
- Dunn, L.A.; Riaz, N.; Fury, M.G.; McBride, S.M.; Michel, L.; Lee, N.Y.; Sherman, E.J.; Baxi, S.S.; Haque, S.S.; Katabi, N.; et al. A Phase 1b Study of Cetuximab and BYL719 (Alpelisib) Concurrent with Intensity Modulated Radiation Therapy in Stage III-IVB Head and Neck Squamous Cell Carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2020, 106, 564–570.
- Zhang, B.; Zhang, Y.; Zhang, J.; Liu, P.; Jiao, B.; Wang, Z.; Ren, R. Focal Adhesion Kinase (FAK) Inhibition Synergizes with KRAS G12C Inhibitors in Treating Cancer through the Regulation of the FAK-YAP Signaling. Adv. Sci. Weinh. Baden-Wurtt. Ger. 2021, 8, e2100250.
- Pifer, P.; Kumar, M.; Yang, L.; Xie, T.; Frederick, M.; Hefner, A.; Beadle, B.M.; Dhawan, A.; Molkentine, D.; Molkentine, J.; et al. Focal Adhesion Kinase Drives Resistance to Therapy in HPV-Negative Head and Neck Squamous Cell Carcinoma in a p53-Dependent Manner. Int. J. Radiat. Oncol. 2022, 112, e12.
- Chan, P.; Han, X.; Zheng, B.; DeRan, M.; Yu, J.; Jarugumilli, G.K.; Deng, H.; Pan, D.; Luo, X.; Wu, X. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol. 2016, 12, 282–289.
- Ramms, D.J.; Raimondi, F.; Arang, N.; Herberg, F.W.; Taylor, S.S.; Gutkind, J.S. Gαs-Protein Kinase A (PKA) Pathway Signalopathies: The Emerging Genetic Landscape and Therapeutic Potential of Human Diseases Driven by Aberrant Gαs-PKA Signaling. Pharmacol. Rev. 2021, 73, 155–197.
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34.
- Campbell, A.P.; Smrcka, A.V. Targeting G protein-coupled receptor signalling by blocking G proteins. Nat. Rev. Drug Discov. 2018, 17, 789–803.
- Wu, V.; Yeerna, H.; Nohata, N.; Chiou, J.; Harismendy, O.; Raimondi, F.; Inoue, A.; Russell, R.B.; Tamayo, P.; Gutkind, J.S. Illuminating the Onco-GPCRome: Novel G protein-coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 2019, 294, 11062–11086.
- Coles, G.L.; Cristea, S.; Webber, J.T.; Levin, R.S.; Moss, S.M.; He, A.; Sangodkar, J.; Hwang, Y.C.; Arand, J.; Drainas, A.P.; et al. Unbiased Proteomic Profiling Uncovers a Targetable GNAS/PKA/PP2A Axis in Small Cell Lung Cancer Stem Cells. Cancer Cell 2020, 38, 129–143.e7.
- Kauko, O.; O’Connor, C.M.; Kulesskiy, E.; Sangodkar, J.; Aakula, A.; Izadmehr, S.; Yetukuri, L.; Yadav, B.; Padzik, A.; Laajala, T.D.; et al. PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci. Transl. Med. 2018, 10, eaaq1093.
- Ho, W.S.; Wang, H.; Maggio, D.; Kovach, J.S.; Zhang, Q.; Song, Q.; Marincola, F.M.; Heiss, J.D.; Gilbert, M.R.; Lu, R.; et al. Pharmacologic inhibition of protein phosphatase-2A achieves durable immune-mediated antitumor activity when combined with PD-1 blockade. Nat. Commun. 2018, 9, 2126.
- Jung, Y.-S.; Park, J.-I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191.
- Morrone, S.; Cheng, Z.; Moon, R.T.; Cong, F.; Xu, W. Crystal structure of a Tankyrase-Axin complex and its implications for Axin turnover and Tankyrase substrate recruitment. Proc. Natl. Acad. Sci. USA 2012, 109, 1500–1505.
- Huang, S.-M.A.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620.
- Azzolin, L.; Zanconato, F.; Bresolin, S.; Forcato, M.; Basso, G.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Role of TAZ as mediator of Wnt signaling. Cell 2012, 151, 1443–1456.
- Juan, W.C.; Hong, W. Targeting the Hippo Signaling Pathway for Tissue Regeneration and Cancer Therapy. Genes 2016, 7, 55.

