1. Mechanisms of YAP/TAZ Activation in Head and Neck Squamous Cell Carcinoma (HNSCC)

1.1. FAT1: A Membrane-Associated Proto-Cadherin Assembling the Hippo Signalome
Although dysregulation of the Hippo pathway occurs frequently in multiple human malignancies, alterations in the canonical core Hippo kinases are rarely observed. In contrast, with an alteration rate of nearly 30%,
FAT1 mutation constitutes one of the most frequent mutations in HNSCC
[1][75]. Inactivating
FAT1 mutations, primarily in the form of homozygous deletions, but also in the form of protein-truncating nonsense mutations, strongly suggest that FAT1 functions as a tumor suppressor
[1][2][3][75,80,81].
Under physiological conditions, FAT1 serves as a membrane-associated scaffold to organize the activated core Hippo signaling complex (signalome). Through loss of function, ectopic expression, and protein interaction experiments, TAOK1/2/3 were demonstrated to represent the upstream kinases triggered by FAT1 to activate the core Hippo complex
[1][75]. In this context, the FAT1 intracellular domain served to assemble the multimeric Hippo core signalome consisting of phospho-MST1 (p-MST1), p-MST2, p-SAV1, p-LATS1, and p-MOB1, culminating in YAP phosphorylation. The degree to which Hippo-mediated YAP phosphorylation is dependent upon FAT1 is underscored by the functional loss of
FAT1, which results in the disassembly of the Hippo signalome with consequent YAP activation, nuclear localization, and activation of YAP-mediated proliferative, survival, and tumorigenic transcriptional programs. These mechanistic observations were validated using cell-based functional assays that demonstrated FAT1 expression abrogated YAP-activated transcription and cell proliferation, which was reversed upon co-expression of a Hippo-insensitive YAP mutant
[1][75]. This
res
earchtudy was the first to mechanistically link
FAT1 disruption with Hippo signaling disruption and uncontrolled YAP activation. Interestingly,
FAT1 mutation has been identified in carcinogen-induced models of HNSCC, potentially linking carcinogen-induced mutation and Hippo pathway perturbation as mediators of HPV-negative HNSCC initiation
[4][82].
FAT2,
FAT3, and
FAT4 are also recurrently altered in human HNSCC and in carcinogen-induced murine models of HNSCC
[4][5][82,83]. However, the biological significance of these alterations remains to be examined
[6][84]. While inactivating
FAT1 mutations are uncommon in HPV-positive HNSCC
[7][85], preliminary analyses suggest that Hippo pathway perturbation and YAP/TAZ activation are also widespread in HPV-positive HNSCC (not shown), thus representing an area of active investigation.
1.2. FAT1-Independent Mechanisms of Oncogenic Hippo Pathway Perturbation in HNSCC
Independent of
FAT1 mutation, YAP activation is a prevalent feature of HNSCC. Epidermal growth factor receptor (EGFR) activation also represents a common event in HNSCC that is correlated with aggressive disease
[8][9][86,87]. Recent studies have described a series of mechanisms through which EGFR and receptor tyrosine kinase downstream signaling cascades via RAS-MAPK and phosphoinositide-3-kinase (PI3K) can drive oncogenic YAP activation.
Amplification and gain of function mutations in
PIK3CA result in the hyperactivation of its gene product PI3K and PI3K signaling pathway activation in HNSCC. PI3K signaling has been shown to induce nuclear YAP localization and YAP-target transcription in response to EGFR or GPCR agonists. Demonstrating strict signaling through PI3K and PDK1, this response was attenuated by PI3K and PDK1 inhibitors and siRNA mediated knockdown. Mechanistically, in cell contact-inhibited conditions, PDK1 binds to MST1 and LATS1 through SAV1, enabling Hippo activation and resulting in YAP phosphorylation and cytoplasmic retention, and cellular growth arrest. In the presence of growth factors or constitutive PI3K signaling, PDK1 is recruited to the plasma membrane, resulting in the dissociation of the MST1:SAV1:LATS1 complex, YAP nuclear accumulation, and cell cycle entry
[10][88].
More recently, EGFR has been shown to activate YAP/TAZ independently of PI3K through the direct phosphorylation of MOB1A in HNSCC cells
[11][44]. EGFR phosphorylates novel MOB1A tyrosine residues in the presence of epidermal growth factor, resulting in reductions in p-LATS1/2 and p-YAP, triggering nuclear translocation of YAP and activation of YAP-mediated transcription programs. Furthermore, inhibition of EGFR in HNSCC cells with erlotinib was shown to be sufficient in abrogating YAP activation and the expression of YAP-mediated transcriptional programs. While further studies are required to delineate the mechanism by which EGFR-mediated MOB1A phosphorylation prevents LATS1 activation, this
res
earchtudy unveiled a novel, therapeutically actionable regulatory signaling node by which multiple receptors and non-receptor tyrosine kinases may converge with the Hippo pathway to control YAP/TAZ activity
[11][44].
Disruption in core Hippo signaling has been shown to induce HNSCC in model systems. Genetic deletion of
Mob1a and
Mob1b in murine lingual epithelia led to rapid squamous cell carcinoma. Consistent with the canonical Hippo core signaling model, knockout of
Mob1a/b in tongue epithelia resulted in diminished LATS1 but not MST1 protein, increased nuclear YAP, and upregulation in YAP target genes
[12][89]. Much remains to be understood about core Hippo signaling and the functions of MOB1 in YAP/TAZ regulation. As illustrated in the setting of
Mob1a/Mob1b knockout in tongue epithelia, knockout of
Yap prevented carcinogenesis induced by
Mob1a/b deletion, but knockout of
Taz in the same context resulted in more aggressive SCC with more invading lesions
[12][89]. While conditional deletion of
Mob1a/b suggests that disruption of these core Hippo complex proteins quickly leads to carcinogenesis,
Mob1a/
b can also contribute to signaling cascades beyond Hippo. For example,
Mob1a and
Mob1b stimulate the activity of non-Hippo kinases including STK38 (NDR1) and STK28L (NDR2)
[13][90]. While NDR1/2 have been implicated in Hippo signaling and reported to phosphorylate YAP
[14][91], NDR1 also serves to attenuate mitogen activated kinase signaling as a negative regulator of MAP3K1/2
[15][92]. Indeed, collectively NDR1/2 exert pleiotropic potentially proto-oncogenic influences on cell cycle progression, apoptosis, stress signaling, and autophagy
[16][93]. Exploring the complex interactions across Hippo and other signaling cascades may clarify seemingly paradoxical effects observed upon the genetic depletion of kinase-regulatory scaffolds, such as
Mob1a/b.
2. Consequences YAP/TAZ Activation in HNSCC
2.1. Cancer Stemness
Chromosome 3q25-26 is a frequently amplified locus in HNSCC that harbors multiple cancer-associated genes including
WWTR1 (TAZ),
PIK3CA, and
SOX2. Co-occurrence of 3q25-26 amplification and
TP53 mutation is associated with poor prognosis in HNSCC
[17][94]. Interestingly, a TAZ:TEAD4 complex has been shown to co-activate
SOX2 transcription in HNSCC. In this context,
SOX2 expression was found sufficient to promote cancer stem cell marker expression, tumor cell self-renewal in vitro, and tumor growth in vivo
[18][95]. Importantly, knockdown of TAZ diminished these phenotypes and exogenous expression of SOX2 upon TAZ knockdown was sufficient to rescue them. Together, these findings show that TAZ- and TEAD4-mediated coactivation of
SOX2 expression is sufficient to induce stemness programs in HNSCC and offer the possibility that TAZ activation may mediate transcriptional programs that lead to the poor prognosis associated with 3q25-26 copy gain. In line with these findings, an independent
res
earchtudy found that TAZ activation promotes migration, invasion, and survival by an epithelial to mesenchymal transition-like program in HNSCC cells, which in turn promotes cancer stem cell maintenance and expansion
[19][96].
The recurrently amplified segment of chromosome 3q also harbors two genes that are frequently co-amplified in HNSCC:
TP63, a key regulator of epidermal cell differentiation and proliferation
[20][97], and
ACTL6A, a subunit of the SWI/SNF ATP-dependent chromatin remodeling complexes
[21][98]. Interestingly, p63 forms a complex with ACTL6A and other SWI/SNF subunits in HNSCC to control a stem-like transcriptional program that enhances the regenerative potential of HNSCC cells in vitro, and promotes a pro-tumorigenic proliferative, undifferentiated cancer cell state in vivo. Importantly, p63 and ACTL6A were found to directly repress transcription of
WWC1 [22][99]. The gene product of
WWC1, Kibra, is a tumor suppressor that promotes Hippo pathway activation
[23][24][100,101], thereby releasing Hippo-mediated inhibition of YAP/TAZ. Indeed, depletion of p63 or ACTL6A, increased Kibra expression, increased p-YAP, depressed nuclear translocation of YAP, and diminished HNSCC regenerative potential in vitro
[22][99].
2.2. Tumor Progression and Poor Prognosis
Progressively increased YAP activation is a feature of oncogenic progression in diverse malignancies
[25][102]. In the squamous epithelia of the upper aerodigestive tract, physiologically nuclear YAP is restricted to the basal epidermis
[1][26][75,103]. The extent and degree of nuclear YAP have been shown to progressively increase with worsening histological severity in oral premalignant lesions and culminate with diffuse, strong nuclear YAP expression in the majority of cells in HNSCC. Furthermore, the frequency of nuclear YAP-positive tumor cells also increases with worsening histological grade in HNSCC
[1][27][75,79]. Array-based transcriptome profiling upon YAP/TAZ knockdown in HNSCC cell lines has elucidated YAP/TAZ-mediated transcription programs in HNSCC. The resultant YAP/TAZ-regulated gene signature, defined as the set of transcripts downregulated upon YAP/TAZ knockdown, showed enrichment for stemness, self-renewal, cell cycle progression, and invasion programs
[27][79]. Consistent with these findings, it has also been shown that nuclear YAP and TAZ in HNSCC are enriched at the tumor invasive front
[28][29][104,105]. In addition, increased nuclear YAP expression in primary tumor specimens has been associated with lymph node metastasis, further suggesting that YAP may play a role in driving invasion and metastatic programs
[29][105]. In line with these findings, TAZ activation was also shown to be a prevalent feature in HNSCC cell lines and an independent prognostic factor for disease-free and overall survival in patients with tongue HNSCC
[30][106].
Whether cancer associated mutations modify prognosis or therapeutic sensitivity are key questions that may shed insight into mechanisms of tumor initiation and progression, and aid in the identification of prognostic, diagnostic, and therapeutic biomarkers. In this regard, the potential roles of YAP/TAZ on prognosis and other clinicopathologic features in HNSCC have been evaluated. YAP copy number alteration (CNA) is observed in 8% of HNSCC. While YAP amplification is associated with increased YAP transcript abundance, augmented YAP expression is often observed in HNSCC in the absence of CNA
[31][107]. Post-translational signaling cascades regulate YAP activation; therefore, YAP activity is likely not directly related to its transcriptional expression levels. Eun and colleagues interrogated the Cancer Genome Atlas (TCGA) database to identify transcripts correlated with YAP transcript expression and CNA in order to develop a gene expression signature for YAP activation
[31][107]. This YAP activation signature was found to stratify prognosis with regard to disease-free survival, disease-specific survival, and overall survival in four independent patient cohorts. Importantly, the YAP activation signature stratified survival after multivariable correction for confounding clinical factors known to be associated with survival (age, tumor classification, nodal classification, TNM staging group, and anatomic site)
[31][107].
Analyses of clinical data-linked, genome-wide databases have identified interactions between PIK3CA and YAP activation in HNSCC and demonstrated that these interactions may be associated with survival outcomes. While neither mutation nor CNA in PIK3CA was associated with recurrence-free survival (RFS), PIK3CA mRNA expression was found to be associated with RFS in TCGA and an independent dataset on single and multivariable analyses
[32][108]. Consistent with mechanistic models in which PIK3CA activation induces downstream YAP dephosphorylation and nuclear translocation to drive YAP-mediated transcription programs, HNSCC tumor samples with high PIK3CA expression displayed lower abundances of p-YAP and PIK3CAhigh tumors expressed transcriptional programs enriched for YAP/TAZ target genes
[32][108]. These provide translational relevance to the mechanistic studies linking PIK3CA activity to YAP/TAZ activation, suggest that the PIK3CA-YAP axis may drive aggressive forms of HNSCC, and provide a strong rationale to target YAP in PIK3CA
high HNSCC.
2.3. Therapeutic Resistance
Combined with radiotherapy and surgical resection, cisplatin-based chemotherapy is a mainstay of curative-intent therapy for HNSCC
[33][109]. Limited evidence suggests that short interfering RNA-mediated YAP-knockdown re-sensitizes cisplatin-resistant HNSCC cell lines to cisplatin. These findings suggest that YAP may be considered a potential therapeutic target for cisplatin-resistant HNSCC
[34][110]. Using a similar line of experimentation, an independent group identified a ribosome-binding protein that mediates the interaction between the ribosome and the endoplasmic reticulum membrane, RRBP1, as a mediator of cisplatin resistance in HNSCC by augmenting YAP expression. In this setting, the use of a putative RRBP1 inhibitor, Radelozid, diminished YAP expression and sensitized HNSCC cells to cisplatin in both in vitro and in vivo assays
[35][111]. In addition, TAZ overexpression has been shown to enhance resistance while YAP or TAZ knockdown sensitizes HNSCC cell lines to cisplatin and fluorouracil
[19][36][96,112].
More recently, precision therapeutic targeting of the MAPK pathway, which is commonly activated in HNSCC and is associated with more aggressive tumor growth, nodal metastasis, and recurrence, has been evaluated in clinical trials
[37][38][39][113,114,115]. In a window-of-opportunity trial in previously untreated HNSCC patients, the small molecule MEK inhibitor trametinib was shown to inhibit the MAPK pathway in 33% of patients, as evaluated by tumor p-ERK abundance, and to result in clinical tumor response rates in 65% of patients
[40][116]. Given that single-modality targeting of MAPK signaling has not shown durable efficacy in a wide variety of tumor types
[41][117], follow up studies have explored mechanisms of trametinib resistance in HNSCC that could lead to multimodal precision therapeutics with durable efficacy. In this regard, trametinib-resistant HNSCC cell lines were generated by culturing cells in sequentially increasing doses of trametinib. The resultant cells demonstrated at least 10
5-fold increased resistance to trametinib and elevated YAP activity as measured by transcriptional reporter assays and upregulation of canonical YAP-target genes. In addition, trametinib-treated patient-derived xenografts that escaped growth inhibition were found to have 15-fold greater abundances of unphosphorylated YAP protein and upregulated YAP-target genes. Accordingly, combining trametinib with verteporfin, which inhibits YAP signaling, showed synergistic effects. These results highlight the potential importance of YAP activation in mediating resistance to MAPK pathway inhibition and suggest YAP inhibition as a potential strategy to enhance the efficacy of MAPK pathway inhibition in patients with HNSCC
[42][118].
3. YAP/TAZ Activation Exposes HNSCC Vulnerabilities and Therapeutic Opportunities
Accumulating evidence demonstrating YAP and TAZ as oncogenic effectors and essential cancer dependencies in HNSCC supports the development of novel oncotherapeutics against YAP/TAZ
[43][44][17,18]. However, therapeutic targeting of transcription factors remains a major challenge. Beyond YAP and TAZ, numerous cancer-associated transcription factors have emerged as promising therapeutic targets
[45][119]. Yet the development of specific small molecule therapeutics against transcription factors has been hampered by hurdles related to the transcription factors not typically possessing enzymatic activities and practical challenges of disrupting protein:protein and protSein:nucleic acid interactions
[45][119].
The requirement for YAP/TAZ to interact with TEAD family DNA binding proteins present an opportunity for its inhibition. An in vitro functional screen in mammalian cells for small molecule inhibitors of YAP:TEAD based transcriptional transactivation identified three porphyrin derivatives protoporphyrin IX, hematoporphyrin, and verteporfin as inhibitors of YAP-induced transcription
[46][120]. Coincidentally, an independent screen identified these same three porphyrins as disruptors of YAP:TEAD interaction in
Drosophila. Further studies validated verteporfin as a strong inhibitor of YAP:TEAD interaction
[46][120]. Verteporfin is an FDA-approved photosensitizer used in photodynamic therapy for the treatment of age-related macular degeneration, suggesting verteporfin exerts cellular effects beyond the inhibition of YAP:TEAD interactions (
Figure 14)
[47][121]. Despite being studied for the past decade as a YAP inhibitor, structural mechanistic details about its interaction with YAP/TAZ remain unclear, and its effects on cellular targets other than YAP/TAZ, including autophagosome inhibition, suggesting that verteporfin constitutes a nonspecific YAP/TAZ inhibitor
[48][122].
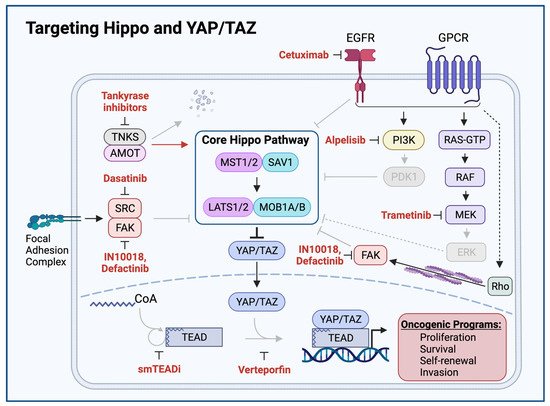
Figure 14. Targeting Hippo and YAP/TAZ. Therapeutic agents promoting Hippo activation or YAP/TAZ inhibition are shown in red. Grey arrows and molecules indicate inhibition of downstream processes. Cetuximab-mediated EGFR inhibition, Alpelisib-mediated PI3K inhibition, Dasatinib-mediated SRC inhibition, Defactinib and IN10018-mediated FAK inhibition, and Trametinib-mediated MEK inhibition relieve Hippo-inhibitory inputs and enable Hippo signaling to inhibit YAP/TAZ. Tankyrase inhibitors prevent proteolytic degradation of AMOT, enabling AMOT to activate Hippo signaling. Small molecule TEAD inhibitors (smTEADi) prevent YAP/TAZ:TEAD interaction by inhibiting TEAD autopalmitoylation. Verteporfin inhibits YAP/TAZ:TEAD interaction
[40][46][49][50][51][52][53][54][55][11,116,120,123,124,125,126,127,128].
Further
res
earchtudy of the YAP/TAZ:TEAD interaction has revealed a unique mechanism through which YAP/TAZ and TEAD family proteins interact to co-activate transcription and has unveiled a novel opportunity for pharmacologic inhibition. YAP/TAZ and TEAD complex formation require an autopalmitoylation event in which TEAD catalyzes the covalent attachment of a palmitate fatty acid to itself. TEAD autopalmitoylation is both necessary for YAP/TAZ:TEAD complex formation and YAP/TAZ-mediated transcriptional co-activation
[56][129]. Small molecule inhibitors of TEAD autopalmitoylation have recently been demonstrated to bind TEAD proteins and prevent TEAD autopalmitoylation, disrupt YAP/TAZ interaction with TEAD proteins, diminish YAP/TAZ:TEAD transcriptional co-activation, downregulate YAP/TAZ:TEAD target transcripts, and inhibit in vivo tumor growth of
NF2-deficient cancer cell lines (
Figure 14)
[50][123]. Whether TEAD autopalmitoylation inhibitors show activity against YAP-activated HNSCC remains an open question and an area of active investigation.
Numerous cellular mechanisms regulate YAP/TAZ activity. Thus, YAP/TAZ inhibition could be achieved through a variety of therapeutic avenues beyond the disruption of YAP/TAZ:TEAD interactions.
TAs described earlier in this review, the GPCR-PKA signaling axis modulates Hippo-YAP/TAZ activity. GPCRs represent one of the most frequently targeted protein classes, with small molecule and peptide drugs designed to target essentially every type of GPCR either approved for clinical use or in clinical development
[57][58][52,130]. Pharmacological inhibition of G
12/13 or G
q/11, or pharmacological activation of G
s could achieve downstream YAP/TAZ inhibition. Attempting to target the Hippo pathway far upstream of YAP/TAZ at plasma membrane bound receptors, however, could result in unforeseen effects
[59][60][131,132].
PKA may represent another therapeutic target in GPCR-PKA-mediated regulation of the Hippo pathway. Interestingly, PKA inhibition via phosphatase PP2A activation has shown antineoplastic activity against models of small cell carcinoma
[61][133]. Given that PP2A drives YAP/TAZ activation, the therapeutic use of PP2A phosphatase activators is unlikely to be beneficial in the treatment of HNSCC. However, drugs that target and inactivate PP2A may prove beneficial in the treatment of HNSCC. PP2A inhibitors are in current development and have shown efficacy against a variety of YAP-activated tumor types
[62][63][134,135].
The interaction of β-catenin and YAP/TAZ also presents a potential therapeutically actionable opportunity. Wnt/β-catenin signaling has independently been associated with tumor initiation and progression and, as a result, multiple inhibitors of this pathway have been developed
[64][136]. Given its role in downregulating β-catenin activity, the β-catenin destruction complex has been a focus for therapeutic intervention with the development of tankyrase inhibitors
[60][61][132,133]. Tankyrase (TNKS) interacts with and degrades the β-catenin destruction complex component Axin via ubiquitin-mediated proteasomal degradation
[65][66][137,138]. Tankyrase inhibitors and other compounds that stabilize Axin have been shown to inhibit both YAP/TAZ and β-catenin activity through the cytoplasmic retention of YAP/TAZ and subsequent restoration of destruction complex activity (
Figure 14)
[44][67][18,139]. In this manner, tankyrase inhibitors promote YAP/TAZ inhibition by the β-catenin destruction complex. More recently, tankyrase inhibitors have been shown to also exert a more direct effect on the Hippo pathway by stabilizing AMOT family proteins, which promote the cytoplasmic retention of YAP/TAZ and prevent YAP/TAZ induced transcriptional programs
[51][68][124,140].