 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sheng Zhu | -- | 1890 | 2022-05-09 08:20:54 | | | |
| 2 | Conner Chen | -3 word(s) | 1887 | 2022-05-09 08:28:18 | | | | |
| 3 | Sheng Zhu | + 407 word(s) | 2294 | 2022-05-09 08:35:07 | | | | |
| 4 | Conner Chen | + 11 word(s) | 2305 | 2022-05-11 08:07:19 | | |
Video Upload Options
Cigarette smoking (CS) is recognized as an independent risk factor for the development of osteoporosis. Clinical studies have illustrated that smokers have significantly lower bone mineral density (BMD) than non-smokers, and cumulative bone loss can increase their lifetime risk of hip fracture by 50%. It has been shown that long-term CS can lead to an imbalance of bone turnover, further contributing to the reduction in bone mass and bone length and increased risk of fractures. Furthermore, chronic consumption of cigarettes has been increasingly linked to impaired muscle function.
1. Indirect Mechanisms
1.1. Body Weight
1.2. Parathyroid Hormone- (PTH-) Vitamin D Axis
1.3. Gonadal Hormones
1.4. Oxidative Stress
2. Direct Mechanisms
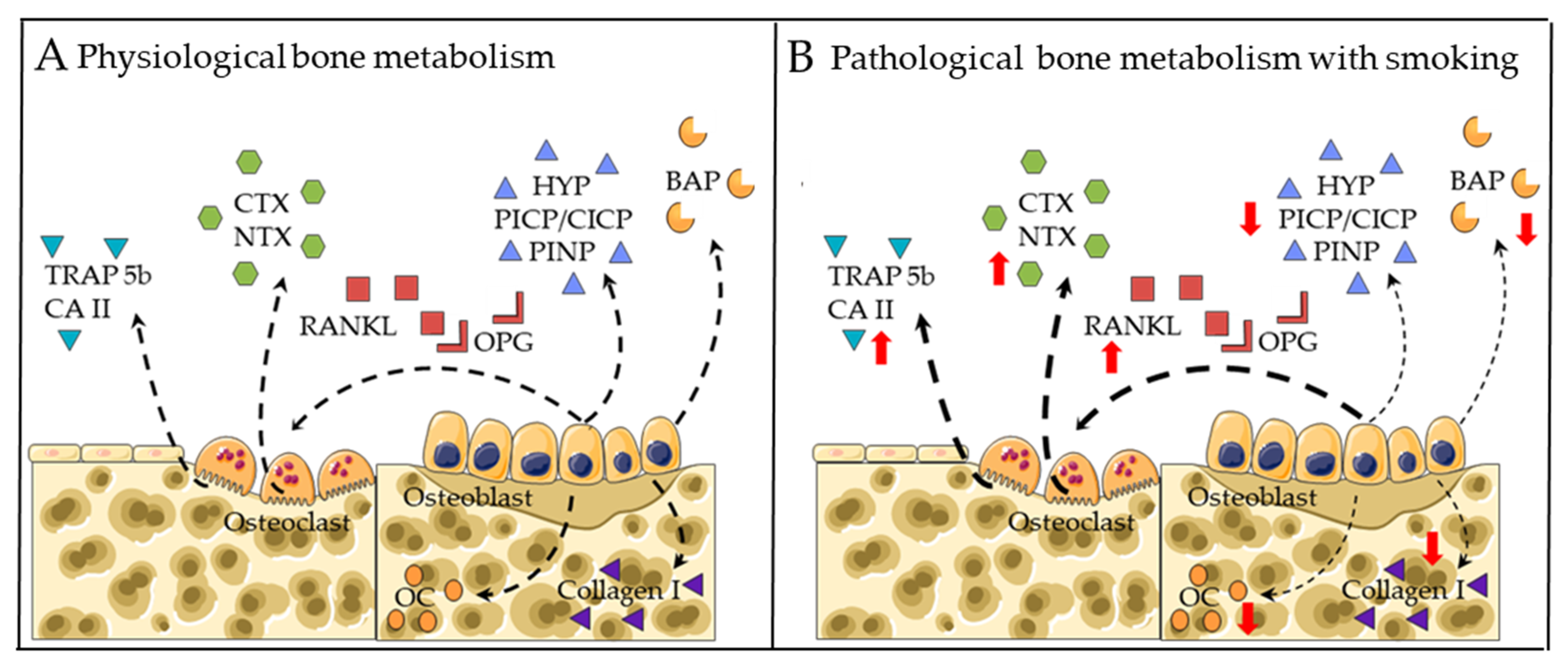
2.1. Markers for Bone Formation
2.2. Markers for Bone Degradation
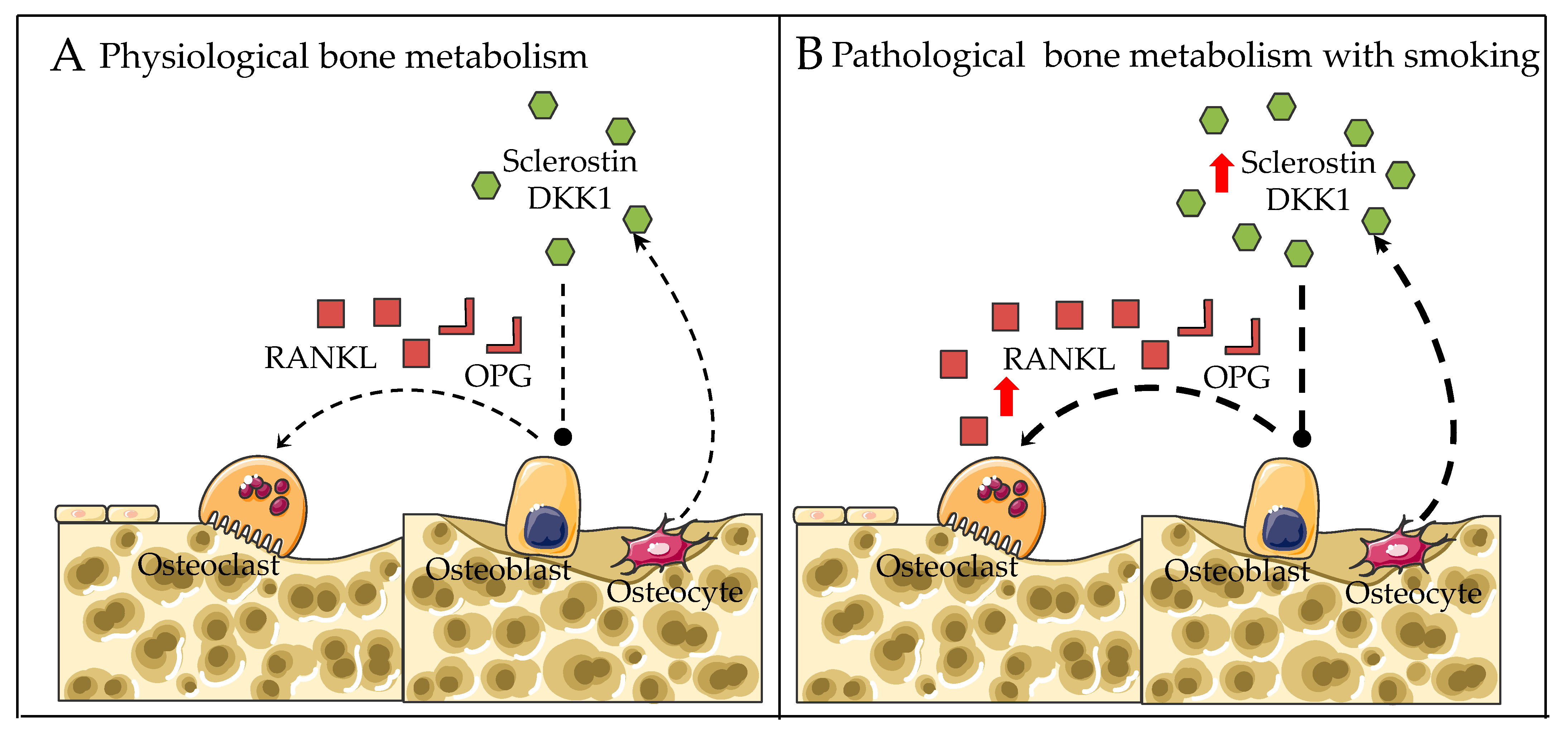
2.3. RANKL-RANK-OPG Pathway
2.4. Wnt/β-catenin Pathway
2.5. Aryl Hydrocarbon Receptor (AhR) Pathway
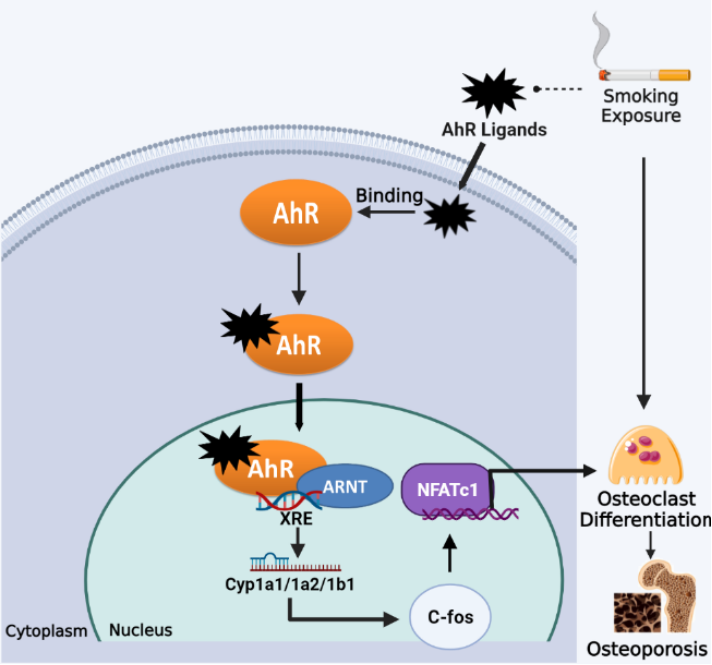
Figure 3. Smoking exposure induces osteoclast differentiation through the AhR pathway, leading to osteoporosis. Dotted lines represent release, arrows represent promotion.
References
- Wang, G.Z.; Zhang, L.; Zhao, X.C.; Gao, S.H.; Qu, L.W.; Yu, H.; Fang, W.F.; Zhou, Y.C.; Liang, F.; Zhang, C.; et al. The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat. Commun. 2019, 10, 1125.
- Xue, J.; Zhao, Q.; Sharma, V.; Nguyen, L.P.; Lee, Y.N.; Pham, K.L.; Edderkaoui, M.; Pandol, S.J.; Park, W.; Habtezion, A. Aryl Hydrocarbon Receptor Ligands in Cigarette Smoke Induce Production of Interleukin-22 to Promote Pancreatic Fibrosis in Models of Chronic Pancreatitis. Gastroenterology 2016, 151, 1206–1217.
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33.
- Yu, H.; Jiang, L.; Wan, B.; Zhang, W.; Yao, L.; Che, T.; Gan, C.; Su, N.; He, J.; Huang, J.; et al. The role of aryl hydrocarbon receptor in bone remodeling. Prog. Biophys. Mol. Biol. 2018, 134, 44–49.
- Iqbal, J.; Sun, L.; Cao, J.; Yuen, T.; Lu, P.; Bab, I.; Leu, N.A.; Srinivasan, S.; Wagage, S.; Hunter, C.A.; et al. Smoke carcinogens cause bone loss through the aryl hydrocarbon receptor and induction of Cyp1 enzymes. Proc. Natl. Acad. Sci. USA 2013, 110, 11115–11120.
- Izawa, T.; Arakaki, R.; Mori, H.; Tsunematsu, T.; Kudo, Y.; Tanaka, E.; Ishimaru, N. The Nuclear Receptor AhR Controls Bone Homeostasis by Regulating Osteoclast Differentiation via the RANK/c-Fos Signaling Axis. J. Immunol. 2016, 197, 4639–4650.
- Eisa, N.H.; Reddy, S.V.; Elmansi, A.M.; Kondrikova, G.; Kondrikov, D.; Shi, X.M.; Novince, C.M.; Hamrick, M.W.; McGee-Lawrence, M.E.; Isales, C.M.; et al. Kynurenine Promotes RANKL-Induced Osteoclastogenesis In Vitro by Activating the Aryl Hydrocarbon Receptor Pathway. Int. J. Mol. Sci. 2020, 21, 7931.
- Lips, P. Vitamin D physiology. Prog. Biophys. Mol. Biol. 2006, 92, 4–8.
- Khazai, N.; Judd, S.E.; Tangpricha, V. Calcium and vitamin D: Skeletal and extraskeletal health. Curr. Rheumatol. Rep. 2008, 10, 110–117.
- Brot, C.; Jorgensen, N.R.; Sorensen, O.H. The influence of smoking on vitamin D status and calcium metabolism. Eur. J. Clin. Nutr. 1999, 53, 920–926.
- Need, A.G.; Kemp, A.; Giles, N.; Morris, H.A.; Horowitz, M.; Nordin, B.E. Relationships between intestinal calcium absorption, serum vitamin D metabolites and smoking in postmenopausal women. Osteoporos. Int. 2002, 13, 83–88.
- Yoon, V.; Maalouf, N.M.; Sakhaee, K. The effects of smoking on bone metabolism. Osteoporos. Int. 2012, 23, 2081–2092.
- Al-Bashaireh, A.M.; Alqudah, O. Comparison of Bone Turnover Markers between Young Adult Male Smokers and Nonsmokers. Cureus 2020, 12, e6782.
- Fujiyoshi, A.; Polgreen, L.E.; Gross, M.D.; Reis, J.P.; Sidney, S.; Jacobs, D.R., Jr. Smoking habits and parathyroid hormone concentrations in young adults: The CARDIA study. Bone Rep. 2016, 5, 104–109.
- Kiel, D.P.; Zhang, Y.; Hannan, M.T.; Anderson, J.J.; Baron, J.A.; Felson, D.T. The effect of smoking at different life stages on bone mineral density in elderly men and women. Osteoporos. Int. 1996, 6, 240–248.
- Prestwood, K.M.; Pilbeam, C.C.; Burleson, J.A.; Woodiel, F.N.; Delmas, P.D.; Deftos, L.J.; Raisz, L.G. The short-term effects of conjugated estrogen on bone turnover in older women. J. Clin. Endocrinol. Metab. 1994, 79, 366–371.
- Mellström, D.; Johnell, O.; Ljunggren, O.; Eriksson, A.L.; Lorentzon, M.; Mallmin, H.; Holmberg, A.; Redlund-Johnell, I.; Orwoll, E.; Ohlsson, C. Free testosterone is an independent predictor of BMD and prevalent fractures in elderly men: MrOS Sweden. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2006, 21, 529–535.
- Shevde, N.K.; Bendixen, A.C.; Dienger, K.M.; Pike, J.W. Estrogens suppress RANK ligand-induced osteoclast differentiation via a stromal cell independent mechanism involving c-Jun repression. Proc. Natl. Acad. Sci. USA 2000, 97, 7829–7834.
- Chen, Q.; Kaji, H.; Kanatani, M.; Sugimoto, T.; Chihara, K. Testosterone increases osteoprotegerin mRNA expression in mouse osteoblast cells. Horm. Metab. Res. 2004, 36, 674–678.
- Bjarnason, N.H.; Christiansen, C. The influence of thinness and smoking on bone loss and response to hormone replacement therapy in early postmenopausal women. J. Clin. Endocrinol. Metab. 2000, 85, 590–596.
- Michnovicz, J.J.; Hershcopf, R.J.; Naganuma, H.; Bradlow, H.L.; Fishman, J. Increased 2-hydroxylation of estradiol as a possible mechanism for the anti-estrogenic effect of cigarette smoking. N. Engl. J. Med. 1986, 315, 1305–1309.
- Barbieri, R.L.; Gochberg, J.; Ryan, K.J. Nicotine, cotinine, and anabasine inhibit aromatase in human trophoblast in vitro. J. Clin. Investig. 1986, 77, 1727–1733.
- Michnovicz, J.J.; Hershcopf, R.J.; Haley, N.J.; Bradlow, H.L.; Fishman, J. Cigarette smoking alters hepatic estrogen metabolism in men: Implications for atherosclerosis. Metabolism 1989, 38, 537–541.
- Cassidenti, D.L.; Vijod, A.G.; Vijod, M.A.; Stanczyk, F.Z.; Lobo, R.A. Short-term effects of smoking on the pharmacokinetic profiles of micronized estradiol in postmenopausal women. Am. J. Obs. Gynecol. 1990, 163, 1953–1960.
- Daniel, M.; Martin, A.D.; Drinkwater, D.T. Cigarette smoking, steroid hormones, and bone mineral density in young women. Calcif. Tissue Int. 1992, 50, 300–305.
- English, K.M.; Pugh, P.J.; Parry, H.; Scutt, N.E.; Channer, K.S.; Jones, T.H. Effect of cigarette smoking on levels of bioavailable testosterone in healthy men. Clin. Sci. 2001, 100, 661–665.
- Wang, W.; Yang, X.; Liang, J.; Liao, M.; Zhang, H.; Qin, X.; Mo, L.; Lv, W.; Mo, Z. Cigarette smoking has a positive and independent effect on testosterone levels. Hormones 2013, 12, 567–577.
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462.
- Callaway, D.A.; Jiang, J.X. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J. Bone Min. Metab. 2015, 33, 359–370.
- Çetin, A.; Muhtaro, S.; Saraymen, R.; Öztürk, A.; Muderris, I.J. Smoking-Induced Bone Defects May Be Due to Oxidative Damage in Postmenopausal Women. Turk. Klin. Tip Bilimleri Derg. 2009, 29, 851–858.
- Zhu, S.; Aspera-Werz, R.H.; Chen, T.; Weng, W.; Braun, B.; Histing, T.; Nüssler, A.K. Maqui berry extract prevents cigarette smoke induced oxidative stress in human osteoblasts in vitro. EXCLI J. 2021, 20, 281–296.
- Aspera-Werz, R.H.; Ehnert, S.; Heid, D.; Zhu, S.; Chen, T.; Braun, B.; Sreekumar, V.; Arnscheidt, C.; Nussler, A.K. Nicotine and Cotinine Inhibit Catalase and Glutathione Reductase Activity Contributing to the Impaired Osteogenesis of SCP-1 Cells Exposed to Cigarette Smoke. Oxid. Med. Cell. Longev. 2018, 2018, 3172480.
- Cusano, N.E. Skeletal Effects of Smoking. Curr. Osteoporos. Rep. 2015, 13, 302–309.
- Kim, B.S.; Kim, S.J.; Kim, H.J.; Lee, S.J.; Park, Y.J.; Lee, J.; You, H.K. Effects of nicotine on proliferation and osteoblast differentiation in human alveolar bone marrow-derived mesenchymal stem cells. Life Sci. 2012, 90, 109–115.
- Marinucci, L.; Balloni, S.; Fettucciari, K.; Bodo, M.; Talesa, V.N.; Antognelli, C. Nicotine induces apoptosis in human osteoblasts via a novel mechanism driven by H2O2 and entailing Glyoxalase 1-dependent MG-H1 accumulation leading to TG2-mediated NF-kB desensitization: Implication for smokers-related osteoporosis. Free Radic. Biol. Med. 2018, 117, 6–17.
- Daffner, S.D.; Waugh, S.; Norman, T.L.; Mukherjee, N.; France, J.C. Nicotine Increases Osteoblast Activity of Induced Bone Marrow Stromal Cells in a Dose-Dependent Manner: An in vitro Cell Culture Experiment. Glob. Spine J. 2012, 2, 153–158.
- Shintcovsk, R.L.; Knop, L.; Tanaka, O.M.; Maruo, H. Nicotine effect on bone remodeling during orthodontic tooth movement: Histological study in rats. Dent. Press J. Orthod. 2014, 19, 96–107.
- Hagiwara, S.; Tsumura, K. Smoking as a risk factor for bone mineral density in the heel of Japanese men. J. Clin. Densitom. 1999, 2, 219–222.
- Law, M.R.; Cheng, R.; Hackshaw, A.K.; Allaway, S.; Hale, A.K. Cigarette smoking, sex hormones and bone density in women. Eur. J. Epidemiol. 1997, 13, 553–558.
- Rudäng, R.; Darelid, A.; Nilsson, M.; Nilsson, S.; Mellström, D.; Ohlsson, C.; Lorentzon, M. Smoking is associated with impaired bone mass development in young adult men: A 5-year longitudinal study. J. Bone Miner. Res. 2012, 27, 2189–2197.
- Ulivieri, F.M.; Silva, B.C.; Sardanelli, F.; Hans, D.; Bilezikian, J.P.; Caudarella, R. Utility of the trabecular bone score (TBS) in secondary osteoporosis. Endocrine 2014, 47, 435–448.
- Sheu, A.; Diamond, T. Diagnostic tests: Bone mineral density: Testing for osteoporosis. Aust. Prescr. 2016, 39, 35.
- Christenson, E.S.; Jiang, X.; Kagan, R.; Schnatz, P. Osteoporosis management in post-menopausal women. Minerva Ginecol. 2012, 64, 181–194.
- Katagiri, T.; Takahashi, N. Regulatory mechanisms of osteoblast and osteoclast differentiation. Oral Dis. 2002, 8, 147–159.
- Matsuo, K.; Irie, N. Osteoclast–osteoblast communication. Arch. Biochem. Biophys. 2008, 473, 201–209.
- Shetty, S.; Kapoor, N.; Bondu, J.D.; Thomas, N.; Paul, T.V. Bone turnover markers: Emerging tool in the management of osteoporosis. Indian J. Endocrinol. Metab. 2016, 20, 846–852.
- Coates, P. Bone turnover markers. Aust. Fam. Physician. 2013, 42, 285–287.
- Nishizawa, Y.; Ohta, H.; Miura, M.; Inaba, M.; Ichimura, S.; Shiraki, M.; Takada, J.; Chaki, O.; Hagino, H.; Fujiwara, S.; et al. Guidelines for the use of bone metabolic markers in the diagnosis and treatment of osteoporosis (2012 edition). J. Bone Miner. Metab. 2013, 31, 1–15.
- Szulc, P. The role of bone turnover markers in monitoring treatment in postmenopausal osteoporosis. Clin. Biochem. 2012, 45, 907–919.
- Ehnert, S.; Aspera-Werz, R.H.; Ihle, C.; Trost, M.; Zirn, B.; Flesch, I.; Schröter, S.; Relja, B.; Nussler, A.K. Smoking dependent alterations in bone formation and inflammation represent major risk factors for complications following total joint arthroplasty. J. Clin. Med. 2019, 8, 406.
- Gurlek, O.; Lappin, D.F.; Buduneli, N. Effects of smoking on salivary C-telopeptide pyridinoline cross-links of type I collagen and osteocalcin levels. Arch. Oral. Biol. 2009, 54, 1099–1104.
- Gao, S.G.; Li, K.H.; Xu, M.; Jiang, W.; Shen, H.; Luo, W.; Xu, W.S.; Tian, J.; Lei, G.H. Bone turnover in passive smoking female rat: Relationships to change in bone mineral density. BMC Musculoskelet. Disord. 2011, 12, 131.
- Oncken, C.; Prestwood, K.; Cooney, J.L.; Unson, C.; Fall, P.; Kulldorff, M.; Raisz, L.G. Effects of smoking cessation or reduction on hormone profiles and bone turnover in postmenopausal women. Nicotine Tob. Res. 2002, 4, 451–458.
- Lehenkari, P.; Hentunen, T.A.; Laitala-Leinonen, T.; Tuukkanen, J.; Väänänen, H.K. Carbonic anhydrase II plays a major role in osteoclast differentiation and bone resorption by effecting the steady state intracellular pH and Ca2+. Exp. Cell Res. 1998, 242, 128–137.
- Jeong, J.W.; Choi, S.H.; Han, M.H.; Kim, G.Y.; Park, C.; Hong, S.H.; Lee, B.J.; Park, E.K.; Kim, S.O.; Leem, S.H.; et al. Protective Effects of Fermented Oyster Extract against RANKL-Induced Osteoclastogenesis through Scavenging ROS Generation in RAW 264.7 Cells. Int. J. Mol. Sci. 2019, 20, 1439.
- Zhu, S.; Haussling, V.; Aspera-Werz, R.H.; Chen, T.; Braun, B.; Weng, W.; Histing, T.; Nussler, A.K. Bisphosphonates Reduce Smoking-Induced Osteoporotic-Like Alterations by Regulating RANKL/OPG in an Osteoblast and Osteoclast Co-Culture Model. Int. J. Mol. Sci. 2020, 22, 53.
- Boyce, B.F.; Xing, L. The RANKL/RANK/OPG pathway. Curr. Osteoporos. Rep. 2007, 5, 98–104.
- Tobeiha, M.; Moghadasian, M.H.; Amin, N.; Jafarnejad, S. RANKL/RANK/OPG Pathway: A Mechanism Involved in Exercise-Induced Bone Remodeling. BioMed Res. Int. 2020, 2020, 6910312.
- Roshandel, D.; Holliday, K.L.; Pye, S.R.; Boonen, S.; Borghs, H.; Vanderschueren, D.; Huhtaniemi, I.T.; Adams, J.E.; Ward, K.A.; Bartfai, G.; et al. Genetic variation in the RANKL/RANK/OPG signaling pathway is associated with bone turnover and bone mineral density in men. J. Bone Min. Res. 2010, 25, 1830–1838.
- Leibbrandt, A.; Penninger, J.M. RANK/RANKL: Regulators of immune responses and bone physiology. Ann. N Y Acad. Sci. 2008, 1143, 123–150.
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 2006, 12, 17–25.
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26.
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem. Biophys. 2008, 473, 139–146.
- Lappin, D.F.; Sherrabeh, S.; Jenkins, W.M.; Macpherson, L.M. Effect of smoking on serum RANKL and OPG in sex, age and clinically matched supportive-therapy periodontitis patients. J. Clin. Periodontol. 2007, 34, 271–277.
- Syed, F.; Khosla, S. Mechanisms of sex steroid effects on bone. Biochem. Biophys. Res. Commun. 2005, 328, 688–696.
- Glass, D.A., 2nd; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell. 2005, 8, 751–764.
- Imamura, A.; Kajiya, H.; Fujisaki, S.; Maeshiba, M.; Yanagi, T.; Kojima, H.; Ohno, J. Three-dimensional spheroids of mesenchymal stem/stromal cells promote osteogenesis by activating stemness and Wnt/β-catenin. Biochem. Biophys. Res. Commun. 2020, 523, 458–464.
- Carrillo-Lopez, N.; Martinez-Arias, L.; Fernandez-Villabrille, S.; Ruiz-Torres, M.P.; Dusso, A.; Cannata-Andia, J.B.; Naves-Diaz, M.; Panizo, S.; European Renal Osteodystrophy, W. Role of the RANK/RANKL/OPG and Wnt/β-Catenin Systems in CKD Bone and Cardiovascular Disorders. Calcif. Tissue Int. 2021, 108, 439–451.
- Ruan, Y.; Kato, H.; Taguchi, Y.; Yamauchi, N.; Umeda, M. Irradiation by high-intensity red light-emitting diode enhances human bone marrow mesenchymal stem cells osteogenic differentiation and mineralization through Wnt/β-catenin signaling pathway. Lasers Med. Sci. 2021, 36, 55–65.
- Choi, H.K.; Kim, G.J.; Yoo, H.S.; Song, D.H.; Chung, K.H.; Lee, K.J.; Koo, Y.T.; An, J.H. Vitamin C Activates Osteoblastogenesis and Inhibits Osteoclastogenesis via Wnt/β-Catenin/ATF4 Signaling Pathways. Nutrients 2019, 11, 506.
- Zhang, Z.; Pan, X.; Chen, M.; Bai, M. Wnt signalling in oral and maxillofacial diseases. Cell Biol. Int. 2022, 46, 34–45.
- Wagner, J.M.; Steubing, Y.; Dadras, M.; Wallner, C.; Lotzien, S.; Huber, J.; Sogorski, A.; Sacher, M.; Reinkemeier, F.; Dittfeld, S.; et al. Wnt3a and ASCs are capable of restoring mineralization in staph aureus-infected primary murine osteoblasts. J. Bone Min. Metab. 2022, 40, 20–28.
- Xu, X.J.; Shen, L.; Yang, Y.P.; Zhu, R.; Shuai, B.; Li, C.G.; Wu, M.X. Serum β -Catenin Levels Associated with the Ratio of RANKL/OPG in Patients with Postmenopausal Osteoporosis. Int. J. Endocrinol. 2013, 2013, 534352.
- Chu, M.; Sun, Z.; Fan, Z.; Yu, D.; Mao, Y.; Guo, Y. Bi-directional regulation functions of lanthanum-substituted layered double hydroxide nanohybrid scaffolds via activating osteogenesis and inhibiting osteoclastogenesis for osteoporotic bone regeneration. Theranostics 2021, 11, 6717–6734.
- Evenepoel, P.; D’haese, P.; Brandenburg, V.J. Sclerostin and DKK1: New players in renal bone and vascular disease. Kidney Int. 2015, 88, 235–240.
- Moysés, R.M.; Schiavi, S.C. Sclerostin, osteocytes, and chronic kidney disease–mineral bone disorder. Semin. Dial. 2015, 28, 578–586.
- Miranda, T.S.; Napimoga, M.H.; Feres, M.; Marins, L.M.; da Cruz, D.F.; da Silva, H.D.P.; Duarte, P.M. Antagonists of Wnt/β-catenin signalling in the periodontitis associated with type 2 diabetes and smoking. J. Clin. Periodontol. 2018, 45, 293–302.


