1. Indirect Mechanisms
1.1. Body Weight
It has been shown that body weight is negatively associated with long-term CS
[1][2][3][34,35,36]. This is probably because nicotine, a major addictive substance in cigarettes, inhibits food ingestion by stimulating the secretion of both dopamine and serotonin
[4][37]. Additionally, CS has been shown to enhance lipid oxidation, contributing to the reduction in body weight
[5][38]. In 2007, Wong et al. provided the underlying mechanisms explaining why long-term CS resulted in a reduction in body weight: (i) weight loss results in a reduction in the mechanical loading on bone, contributing to a decreased stimulus for osteogenic differentiation; (ii) CS cause a reduction in adipose tissue by inhibiting the conversation of androgen to estrogen; and (iii) CS cause a reduction in leptin levels, which has been known to enhance bone mass
[6][12].
1.2. Parathyroid Hormone- (PTH-) Vitamin D Axis
The PTH-vitamin D axis exerts crucial functions in maintaining bone density and calcium hemostasis
[7][39]. PTH has a regulatory function in regulating serum calcium levels by bone and kidney reabsorption, while vitamin D can modulate the absorption of calcium from the intestine
[8][9][40,41]. Several studies have indicated that smoking downregulates vitamin D serum levels
[10][11][42,43] and inhibits the production of PTH
[12][44]. However, the influence of CS on the inhibition of the production of PTH remains controversial
[13][14][45,46]. These inconsistencies may be due to confounding effects of body weight, alcohol consumption, medication use and calcium and vitamin D supplementation
[10][15][42,47].
1.3. Gonadal Hormones
Estrogen and testosterone exert essential functions in modulating bone remodeling
[16][17][48,49]. It has been shown that estrogen inhibits osteoclast differentiation and bone resorption
[18][50], while testosterone promotes proliferation and osteogenic differentiation
[19][51]. In females, CS causes a reduction in estrogen levels by altering estrogen metabolism
[20][21][52,53]. Several explanations behind this phenomenon are as follows: (i) nicotine and cotinine suppress aromatase enzyme activity, contributing to a reduction in estrogen levels
[22][54]; (ii) CS promotes estradiol to decompose into 2-methoxyestrone in the liver
[23][55]; and (iii) CS enhances serum hormone-binding globulin levels, leading to a reduction in free estradiol levels in the blood
[24][25][56,57]. In males, current findings are contradictory. Some research indicated testosterone levels were comparable between smokers and nonsmokers, whereas other research showed that smokers had higher testosterone levels than nonsmokers
[26][27][58,59]. Like females, the potential mechanism of aromatase inhibition was involved in males
[12][44].
1.4. Oxidative Stress
2.1.4. Oxidative Stress
The presence of reactive oxygen species (ROS) in bone can influence resident cell behavior, extra-cellular matrix composition and tissue architecture. CS is related to high levels of ROS, which improves osteoclast activity and suppresses osteoblast activity, leading to a reduction in bone mass
[28][29][60,61], and maqui berry extract, a fruit extract rich in anthocyanins, is effective against the effects of CSE on osteoblasts. A clinical study demonstrated that smoking increased levels of oxidative stress
[30][62]. Consistently, Zhu and colleagues observed that cigarette smoke extract (CSE) significantly enhanced ROS and nitrotyrosine levels in human osteoblasts
[31][63]. Probing potential molecular mechanisms, Aspera-Werz et al.reported that Nrf2 directly modulates antioxidant defense systems in human mesenchymal stem cells exposed to CSE
[32][64], and resveratrol could decrease ROS level generated by CSE to protect the primary cilia integrity in human bone mesenchymal stem cells.
2. Direct Mechanisms
CS has been found to exert a direct role in bone tissue through binding to several receptors, including nicotinic acetylcholine receptors and androgen receptors in osteoblasts and aryl hydrocarbon receptors in osteoclasts
[33][11]. Nicotine, a major addictive substance in cigarettes, binds to nicotinic receptors in osteoblasts
[34][65]. However, the role of nicotine in bone metabolism has been controversial. Marinucci et al. observed that nicotine negatively contributed to the impaired osteogenic differentiation of mesenchymal stem cells (MSCs)
[35][66]. On the contrary, Daffner and colleagues demonstrated that nicotine could even promote MSC proliferation and osteogenic differentiation at concentrations lower than its serum concentration in smokers
[36][67]. Aspera-Werz et al. showed that the osteogenic differentiation of MSCs was not largely altered when the cells were exposed to nicotine at concentrations similar to its serum levels in smokers
[32][64]. Similarly, Shintcovsk and colleagues demonstrated that nicotine had no influence on osteoclastogenesis
[37][68]. Therefore, nicotine may not be a major compound responsible for impaired bone metabolism in bone cells.
Many studies have reported the negative relationship between CS and bone mass
[38][39][40][69,70,71]. However, early diagnosis of impaired bone metabolism remains a great challenge. Due to the lack of obvious symptoms in the early stages of CS-induced osteoporosis, the bone metabolism of smokers has been impaired for a long period of time when they start to feel back pain or suffer from fractures
[41][42][72,73]. Actually, after reaching peak bone mass around the age of 30 in adults, bone loss occurs at varying rates, and smoking significantly accelerates this process. Hence, it is highly desirable to detect the alternations of bone metabolism in the early stages to prevent the development of osteoporosis
[43][4].
Several factors secreted into the blood by osteoblasts and osteoclasts regulate osteoblast-medicated bone formation and osteoclast-mediated bone resorption
[44][45][74,75]. These serum markers represent a crucial complementary diagnostic tool to bone mineral density
[46][76]. In general, monitoring these secreted markers could be helpful in determining the alternations in bone metabolism before the manifestation of osteoporosis.
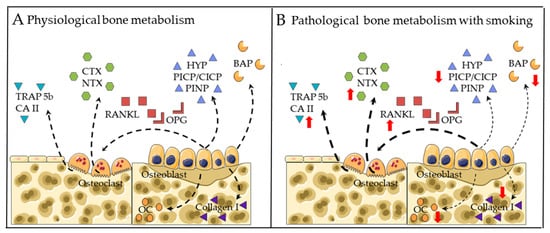
Figure 1 summarizes the direct effects of CS on bone metabolism.
Figure 1. Established gene markers for bone metabolism in (A) physiological conditions and (B) CS exposure conditions. Bone resorption markers include: tartrate-resistant acid phosphatase isoform 5b (TRAP5b), carbonic anhydrate II (CA II), C-terminal telopeptide (CTX) and N-terminal telopeptide (NTX). Regulators of osteoclastogenesis include: receptor activator of nuclear factor kappa B ligand (RANKL) and osteoprotegerin (OPG). Bone formation markers include: osteocalcin (OC), collagen I, bone-specific alkaline phosphatase (BAP), hydroxyproline (HYP) and type I collagen N- and C-terminal propeptides (PINP and PICP/CICP). Dotted arrows represent expression. Red arrows represent altered expression (up or down).
2.1. Markers for Bone Formation
Hydroxyproline (HYP) and type I collagen N- and C-terminal propeptides (PINP and PICP/CICP) in blood are commonly used to evaluate bone formation
[47][48][49][77,78,79]. Ehnert and colleagues demonstrated that following total joint replacement, heavy smokers had a greater risk of prolonged hospital stay due to reduced levels of CICP as compared to nonsmokers
[50][80]. Additionally, there are alternative markers involved in osteogenesis. Gürlek et al. reported that smoking significantly decreased salivary levels of osteocalcin (OC, later stage of osteogenic differentiation)
[51][81]. Supporting these data, in a rat model, Gao et al. reported that chronic exposure to smoking significantly decreases levels of bone-specific alkaline phosphatase (BAP, early stage of osteogenic differentiation) and OC, resulting in osteoporotic bone
[52][82].
2.2. Markers for Bone Degradation
C-terminal telopeptide (CTX) and N-terminal telopeptide (NTX) in blood are commonly used to represent collagen-I degradation
[48][49][78,79]. Oncken et al. demonstrated that smoking cessation contributed to a reduction in NTX levels in postmenopausal women
[53][83]. In addition, there are alternative markers involved in osteoblastogensis.
Our Anprevious in vitro study revealed that chronic exposure to CSE-enhanced carbonic anhydrate II (CA II, early stage of osteoclastic differentiation
[54][84]) and tartrate-resistant acid phosphatase 5b (TRAP 5b, later stage of osteoclastic differentiation
[55][85]) activities in a bone co-culture system, resulting in an osteoporotic microenvironment
[56][86].
Although these markers can represent bone metabolic alternations, the underlying mechanisms by which CS induces an osteoporotic bone environment are not yet fully understood. Understanding the regulatory mechanisms in bone cells impaired by CS will enable reusearchers to obtain insights into the development of CS-induced osteoporotic bones and screen relevant preventive treatment strategies for smokers with osteoporotic bones.
2.3. RANKL-RANK-OPG Pathway
It has been shown that the receptor activator of nuclear factor-kappa B ligand (RANKL)-RANK-osteoprotegerin (OPG) (RANKL-RANK-OPG) pathway exerts a crucial role in osteoclast differentiation and bone resorption
[57][58][59][87,88,89]. Leibbrandt and colleagues revealed that osteoclasts failed to be differentiated in a RANKL and RANK knockout mouse model, indicating RANKL is crucial for osteoclastic differentiation and activation
[60][90]. RANKL, a membrane protein, is synthesized and released by osteoblasts, which is capable of binding to the RANK receptor located on osteoclasts, activating osteoclastogenesis
[61][91]. As a decoy receptor for RANKL, OPG suppresses osteoclastogenesis by binding to RANKL
[62][92]. Hence, the balance between RANKL and OPG exerts a vital role in regulating bone metabolism
[63][93].
Currently, few studies have been conducted regarding the association between CS and the RNKL-RANK-OPG pathway. In terms of human studies, researchers have demonstrated that smoking significantly reduced OPG levels, leading to an increased RANKL/OPG ratio
[13][64][45,94]. Consistently, in an in vitro bone co-culture system,
aour previous research showed that chronic exposure to CSE enhanced osteoclast function via upregulation of the RANKL/OPG ratio, resulting in an osteoporotic microenvironment, and bisphosphonates can be used to counteract the effects of CSE on the RANKL/OPG pathway
[56][86].
Several studies have shown the RANKL-RANK-OPG pathway is also involved in indirect pathophysiological mechanisms (PTH-vitamin D axis and gonadal hormones), which makes this pathway of great therapeutic interest
[60][90]. Moreover, this pathway, in interaction with several factors, such as prostaglandin E2 and interleukins, further affects bone metabolism
[60][90]. For instance, estrogen and androgen could enhance OPG levels in osteoblasts and downregulate the RANKL/OPG ratio, further inhibiting osteoclast differentiation
[65][95].
2.4. Wnt/β-catenin Pathway
The canonical Wnt/β-catenin signaling pathway may be one potential mechanism that is involved in the regulation of RANKL/OPG balance. Glass et al. reported that the activation of the Wnt/β-catenin pathway in osteoblasts can downregulate the RANKL/OPG ratio, leading to compromised osteoclast differentiation and bone resorption
[66][96]. Apart from suppressing osteoclastogenesis, it also enhances the osteogenic differentiation of MSCs
[67][97], bone formation
[68][98] and mineralization
[69][99]. Therefore, the Wnt/β-catenin pathway appears to exert a dual role in bone remodeling, enhancing osteogenesis on the one hand and suppressing osteoclastogenesis on the other hand. Understanding and characterizing the Wnt/β-catenin pathway in detail is believed to be critical to guiding the interaction between osteoblast and osteoclast
[70][100].
Wnt ligands, which secrete glycoproteins, are engaged in activating the Wnt/β-catenin canonical pathway
[71][101]. When Wnt ligands (Wnt 3a and 5a) bind with receptor complexes, including Frizzled and LRP5/6, the Wnt/β-catenin pathway is activated to accumulate β-catenin in the cytoplasm of osteoblasts
[72][102]. Then, the accumulated β-catenin transfers into the nucleus to regulate the transcription of osteogenic-related genes such as Osterix,
RUNX2 and
OPG [73][103]. Eventually, the accumulated OPG binds with RANKL and prevents it from integrating with RANKL located on osteoclasts, inhibiting osteoclastogenesis
[74][104]. Additionally, sclerostin and Dickkopf-1 (DKK1) are potent targets, blocking the canonical Wnt signaling pathway
[75][105]. DKK1 is expressed by osteocytes and mature osteoblast, while sclerostin is expressed by osteocytes
[76][106].
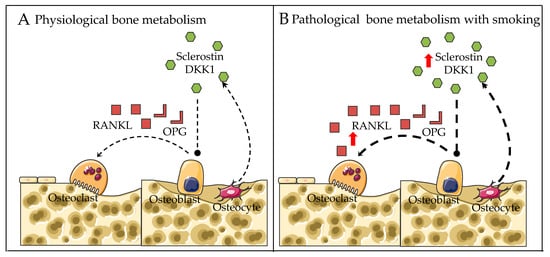
To date, few studies have been carried out showing the association between CS and inhibitors of Wnt signaling. Jorde et al. reported that smokers had higher levels of DKK1 and lower levels of bone formation (PINP) when compared to nonsmokers
[7][39]. Supporting these data, Miranda et al. revealed that smoking could enhance the levels of DKK1 and sclerostin in patients with periodontitis
[77][107]. Thus, the inhibition of Wnt signaling by CS may result in the upregulation of the RANKL/OPG ratio, leading to excessive osteoclast differentiation and bone resorption (
Figure 2).
Figure 2. Proposed regulatory mechanisms for bone metabolism in (A) physiological conditions and (B) smoking exposure conditions. Dotted arrows represent expression. Red arrows represent altered expression (up or down).
2.5. Aryl Hydrocarbon Receptor (AhR) Pathway
Smoking exposure potently activates the cellular Aryl hydrocarbon receptor (AhR) [78], as cigarette smoke contains a large number of exogenous ligands for AhR, such as dioxin-like derivatives represented by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and a variety of polycyclic aromatic hydrocarbon (PAH)-based compounds such as benzo(a)pyrene (BaP) [79]. The AhR is one of the ligand-activated transcription factors for sensing environmental pollutants, which stays in an inactive form in the cytoplasm. Upon binding to exogenous ligands from smoke, the conformation of the AhR is changed, which enables AhR translocation into the nucleus, where it heterodimerizes with the aromatic hydrocarbon receptor nuclear transfer protein (ARNT) and activates xenobiotic response elements (XRE), to the initial transcription of target genes (CYP1a1/CYP1a2/CYP1b1) and leads to different toxicological and pathological effects [79][80]. Recent studies have shown that endogenous activation of the AhR has multiple roles in the regulation of bone remodeling and its associated signaling pathways, which may be an essential mechanism for smoking-induced osteoporosis [81].
Jameel et al. reported that BaP and TCDD in cigarette smoke caused AhR activation and a significant increase in CYP1A1 and CYP1A2 expression, resulting in enhanced OC differentiation and compromised bone mass in wild-type mice but not in AhR-/- mice [33]. Moreover, TCDD administration was unable to stimulate osteoclastogenesis in bone marrow macrophages (BMMs) from CYP1A1/CYP1A2 double knockout and CYP1A1/CYP1A2/CYP1B1 triple knockout mice, unlike in BMM from wild-type mice [33]. These results suggest that AhR-induced CYP1 expression mediates at least in part the activation of osteoclastogenesis by AhR ligands from cigarette smoke. Further studies found that RANKL-stimulated osteoclastogenic signaling (e.g., phosphorylation of Akt, MAPK and NF-κB) was impaired in AhR-/- BMMs [82]. Furthermore, the AhR was found to be activated by RANKL at an earlier stage than signature osteoclastic genes (e.g., CSTK and NFATc1) in pre-osteoclasts, suggesting a regulatory role of the AhR in OC differentiation. BaP was shown to induce higher levels of c-Fos in RANKL-stimulated BMMs, but c-Fos was not induced by BaP or RANKL in AhR-/- BMMs [82]. Consistently, the AhR activation in BMMs was also found to lead to significant upregulation of c-Fos and NFATc1 expression, thereby initiating osteoclastic differentiation [83]. Taken together, the AhR pathway mediates excessive osteoclastic differentiation and bone resorption caused by smoking exposure, which could be a potential target for the prevention of smoking-induced osteoporosis (Figure 3).
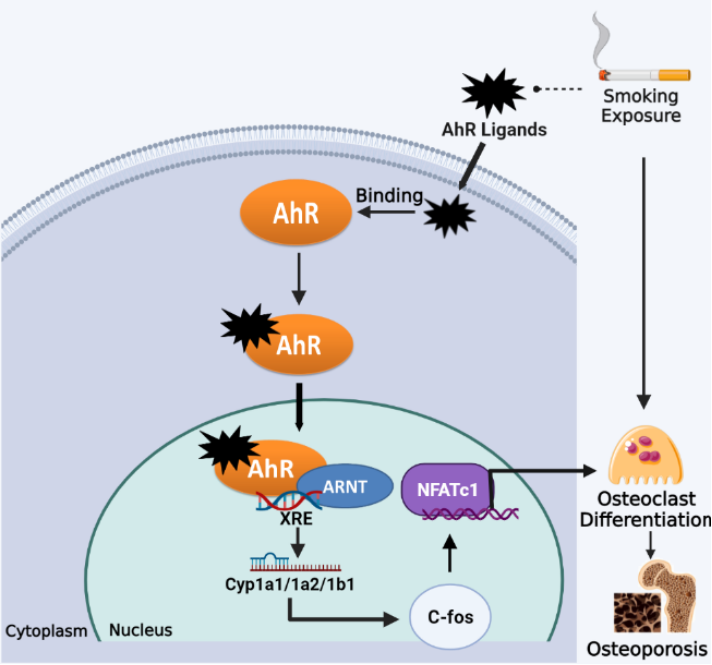
Figure 3. Smoking exposure induces osteoclast differentiation through the AhR pathway, leading to osteoporosis. Dotted lines represent release, arrows represent promotion.