Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Xu, J.; Wang, A.; , .; Bao, J.; Yakubu Saddeeq, A.; Lu, G.; Wang, Z. SNARE Genes in Brassica napus. Encyclopedia. Available online: https://encyclopedia.pub/entry/22584 (accessed on 26 July 2026).
Xu J, Wang A, , Bao J, Yakubu Saddeeq A, Lu G, et al. SNARE Genes in Brassica napus. Encyclopedia. Available at: https://encyclopedia.pub/entry/22584. Accessed July 26, 2026.
Xu, Jing, Airong Wang, , Jiandong Bao, Abubakar Yakubu Saddeeq, Guodong Lu, Zonghua Wang. "SNARE Genes in Brassica napus" Encyclopedia, https://encyclopedia.pub/entry/22584 (accessed July 26, 2026).
Xu, J., Wang, A., , ., Bao, J., Yakubu Saddeeq, A., Lu, G., & Wang, Z. (2022, May 03). SNARE Genes in Brassica napus. In Encyclopedia. https://encyclopedia.pub/entry/22584
Xu, Jing, et al. "SNARE Genes in Brassica napus." Encyclopedia. Web. 03 May, 2022.
Copy Citation
SNAREs (soluble N-ethylmaleimide-sensitive factor attachment protein receptors) are central components that drive membrane fusion events during exocytosis and endocytosis and play important roles in the different biological processes of plants.
Brassica napus
SNARE
gene family
Sclerotinia sclerotiorum
expression profile
hormone stimuli
1. Introduction
Vesicle trafficking is essential for diverse biological processes including cell polarity, growth, development, and adaptation [1,2,3,4,5]. The cargo exchange of trafficking vesicles promotes the vesicle-mediated communication among eukaryotic cells through the exocytic and endocytic pathways. These exocytic and endocytic processes are dependent on the targeted membrane fusion of vesicles that delivers membranes, proteins, and soluble cargos between subcellular membranous compartments and the plasma membrane [6]. This membrane fusion mechanism is highly conserved in all eukaryotes [7] and the central components driving the actual membrane fusion events are a set of proteins dubbed as SNAREs (soluble N-ethylmaleimide-sensitive factor attachment protein receptors) [8].
SNAREs can be classified into v-SNAREs (associated with transport vesicles) and t-SNAREs (associated with the target compartments) [9]. Considering that some SNARE proteins may have multiple functions, another classification method is generally accepted: Qa-, Qb-, Qc-, Qbc-, and R-SNAREs according to the central amino acid present in the hydrophobic heptad repeats of the proteins [10]. The fusion of vesicles with their target membrane is driven by a cluster of four coiled-coil helices, termed Qa, Qb, Qc, and R, each of which is contributed by three or four individual SNARE proteins (a single Qbc-SNARE protein carries two SNARE helices: Qb and Qc) [11]. The specific interaction between v-(R)SNARE and a cognate set of t-(Q)SNAREs is an important part of the mechanisms that partly influence the accuracy of the transport. SNARE proteins form a superfamily of diverse proteins with at least 64 members in Arabidopsis thaliana [11], 60 members in rice [11], 63 members in tomato [12], 69 members in Populus trichocarpa [13], and 173 members in wheat [14]. Compared to other eukaryotes, plants so far have the highest number of identified SNAREs; Homo sapiens has 38 SNAREs [15], Drosophila melanogaster has 26 [16], and there are between 21 and 25 of them in Saccharomyces cerevisiae [17,18]. Sansebastiano and Piro were of the opinion that this increase in the number of SNAREs in plants was a result of gene expansion of partially redundant genes in conserved subfamilies and not the evolution of new isoforms [13]. The expansion of SNARE encoding genes in plants implies the importance of this superfamily during the growth and development of the plants, as well as for biotic and abiotic stress responses [19].
During the last 20 years, evidence for the diverse functions of SNAREs at multiple stages of plant development rapidly accumulated. At the cellular level, SNARE proteins express in diverse organelles such as the plasma membrane, ER, transport vesicles, Golgi apparatus, and trans-Golgi network (TGN). The proteins are previously reported to mediate the processes of vacuole biogenesis, vacuolar transport, vesicle fusion, secretion, cell growth, and ion homeostasis. AtVAMP721 interacts with the potassium channels AtKAT1 and AtKC1 to maintain the currency of the K+ channels in A. thaliana [20]. Members of AtSYP4 (AtSYP41, AtSYP42, AtSYP43) localize on the same TGN compartment and maintain the morphology of both the Golgi apparatus and TGN [21]. The homodimer form of the ER-localized R-SNARE protein AtSEC22 plays a major role during anterograde and retrograde transports by promoting efficient membrane fusion and assisting in the assembly of higher-order complexes. Furthermore, the Qc-SNARE AtBET12 together with the Qb-SNARE AtMEMB12 negatively regulates the secretion of pathogenesis-related protein 1(PR1) in A. thaliana [22].
At the tissue level, several types of SNAREs were reported to play vital roles in root growth, pollen tube growth, and seed maturation. For example, the membrane-localized Qa-SNARE AtKNOLLE (AtSYP111) is highly expressed in organs containing dividing cells and is specifically involved in cytokinetic vesicle fusion [23]. AtSYP123 is expressed and accumulated in the cells present at the tip region of root hairs during root development, while AtSYP124, AtSYP125, and AtSYP131 only express in pollen and are involved in pollen tube growth [24].
At the whole-plant level, SNAREs are mainly activated in response to stresses such as drought/osmotic stress, high salinity, abscisic acid (ABA)-induced stress, and pathogen stimuli. For example, AtSYP121 is involved in ABA-dependent drought stress in tobacco and non-host resistance against powdery mildew as well as oomycete attack in A. thaliana [2,23,25]. As a paralog of AtSYP121, AtSYP122 is phosphorylated in response to the elicitor flagellin [26] and has redundant functions with AtSYP121 in plant immunity and general secretion events [27,28]. Another Qa-SNARE, SYP132 also plays roles in bacterial defense and symbiosome definition in Nicotiana benthamiana and Medicago truncatula, respectively [29,30]. AtSYP61 plays an important role in osmotic stress tolerance and the ABA-dependent regulation of stomatal responses [31]. ShNPSNl11 plays a positive role in defense activation and host resistance to Oidium neolycopersici in tomato [25].
Brassica napus is a major oil crop in temperate regions of the world. It belongs to the family Brassicaceae. The amphidiploid B. napus (2n = 38, AACC) was formed as a hybrid between progenitors of B. rapa (2n = 20, AA) and B. oleracea (2n = 18, CC) ~7500 years ago [32], both of which underwent whole-genome triplication [33,34]. More ancient polyploidization events [35,36] along with the recent hybridization and subsequent gene loss shaped the B. napus genome and determined the size of the entire gene complement ~100,000 genes as well as the individual gene families [37]. This evolutionary process and the close relationship of B. napus with A. thaliana make B. napus an ideal material for gene family evolutionary research. However, the number, nature, general relationships, and functions of the various SNARE proteins present in B. napus remain in the dark. Therefore, in this study, we first used an in silico approach to carry out global identification of the members of the SNARE family in B. napus and then systematically analyze their structural similarities, evolutionary relationships, and transcriptional profiles under the influences of the necrotrophic fungus Sclerotinia sclerotiorum, oxalic acid (OA), methyl jasmonate (MeJA), salicylic acid (SA), and abscisic acid (ABA).
2 Identification of SNARE Genes in B. napus
To identify all members of the SNARE family in B. napus, three methods including Pfam analysis, conserved domains search, and orthologous sequence BLAST were used. A total of 237 BnaSNAREs were identified. Of these genes, six (6) couples sharing 100% identity on the amino acid level but different nucleotide sequences were considered to be different BnaSNAREs. All candidate BnaSNAREs were named according to their best hit in Arabidopsis. Each gene name starts with an abbreviation for the species name B. napus (Bna), followed by the name of the most prominent Arabidopsis gene from this subclade (e.g., “BnaSYP122” for AtSYP122-like genes, “BnaSEC20” for AtSEC20-like genes, “BnaYKT61” for AtYKT61-like genes). Exceptions are BnaSNAP33s, BnaSNAP30s, and BnaSNAP29s which are named according to their protein molecular weight (e.g., “BnaSNAP31” represents the protein molecular weight is approximate 31 kDa). Genes on different chromosomes belonging to the same subclade were consecutively numbered according to their chromosome number from low-to-high values (e.g., four KNOLLE-like genes BnaC05g06210D, BnaA06g04950D, and BnaA08g26870D, BnaC08g13620D were named separately as “BnaKNOLLEa”, “BnaKNOLLEb”, “BnaKNOLLEc”, and “BnaKNOLLEd”). In the case of SYP4s, our phylogenies did not provide clear orthologous relationships among SYP41s, SYP42s, and SYP43s genes from B. napus and Arabidopsis. We therefore named the SYP4 subclade genes of B. napus as BnaSYP44, BnaSYP45, and BnaSYP46, taking up the current code of Arabidopsis. A similar strategy was adopted to name USE1, SFT1, MEMB1, and YKT62-like genes. VAMP724, VAMP726, and VAMP728 genes were similar to the case of BnaSYP4s, but the strategy did not fit this case because the codes in Arabidopsis are up to eight (VAMP728). “Slash” was rather used in naming these genes (e.g., “BnaVAMP724/6/8a”, “BnaVAMP724/6/8b”).
Almost all of the identified SNAREs in B. napus showed the same conserved domain with their respective orthologs in Arabidopsis according to NCBI batch CD search, except for the 11 SYP6-like proteins, which just contain 1 N-terminal syntaxin-6 (PF09177) domain but lost a C-terminal SNARE domain. Furthermore, of these 11 SYP6-like proteins from B. napus, 3 orthologs from Arabidopsis were not previously identified. Considering the fact that the conserved N-terminal syntaxin-6 domain is unique to the SNARE family, we believe that the 11 genes belong to the SNARE family and we classified them as SYN-sub-family.
The BnaSNARE proteins have varying physicochemical characteristics. Isoelectric points (pIs) of the proteins are between 4.44 and 11.84, and their molecular weights (MWs) range from 9.02 to 120.51 kDa. Exceptions are BnaSYP31d with 7.15 kDa, and both BnaSYP112e and BnaSYP112f have MW of 8.05 kDa, which fall below the range; BnaVTI11e, however, has an MW of 184.07 kDa which falls above the stated range. Subcellular localization prediction for the BnaSNARE proteins indicated that they are localized at the plasma membrane, ER, Golgi, vacuole, and a small group were located in the cytoplasm, mitochondrion, and nucleus.
3 BnaSNAREs Belong to Well-Defined Subfamilies That Were Correlated to Their Gene Structures and Conserved Motifs
A maximum-likelihood phylogenetic tree of all the SNARE genes from A. thaliana and B. napus shows that the B. napus genome retains all the orthologs of A. thaliana SNAREs and the gene phylogeny roughly followed species phylogeny. In several subclades, one SNARE in A. thaliana is closely related to a double of two B. napus homologs (e.g., SFT1, USE1 Figure 1 and Table S4) which is consistent with the chromosome multiples of B. napus and A. thaliana as B. napus is heterotetraploid while A. thaliana is diploid. In many subclades, SNARE homologs in B. napus are significantly expanded compared to those in A. thaliana. Measured from the total point of view, the number of SNAREs in B. napus is much more than double of those in A. thaliana, in fact, nearly four times (e.g., TYN1, SEC20, SYP5 subclades Figure 1 and Table S1). The topology in BnaSYP12, BnaYKT6, BnaSYP6, and BnaVAMP72 subclades (Figure 1 and Table S1) is more complex, suggesting multiple duplication events, before and/or after polyploidization of B. napus. The BnaSNARE proteins displayed the same five groups described previously (Q (a-, b-, c-, bc-) and R) in A. thaliana and presented a similar proportion of members compared to A. thaliana (Table S5). Therefore, Qa-, Qb-, and Qc-BnaSNAREs are composed of 69, 44, and 37 (+11) genes, respectively. Qbc-BnaSNAREs have 10 members and R-BnaSNAREs have 65 members.
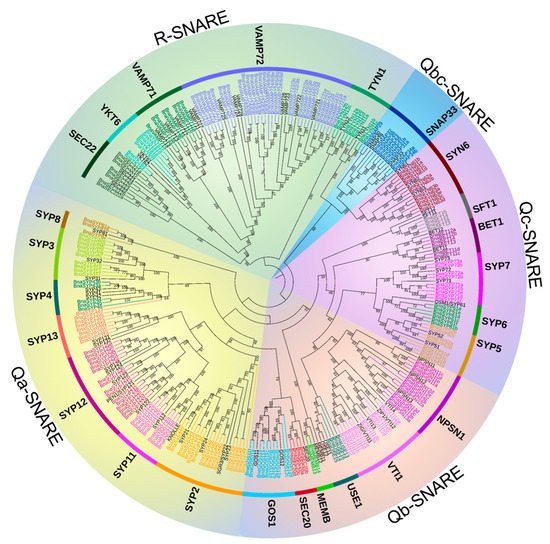
Figure 1. Maximum-likelihood phylogeny of SNARE proteins from B. napus and A. thaliana. A phylogenetic tree of SNARE proteins from B. napus and A. thaliana was constructed using IQ-TREE. The colored B. napus genes are subclade-specific, whereas A. thaliana genes were in black. Subfamilies were indicated using A. thaliana gene names, and sub-family names according to priority rule [11] were shown in brackets if different from the A. thaliana gene names. Despite the absence in other research, the A. thaliana genes SYN61, SYN62, and SYN63 and their orthologous in B. napus were included in the phylogeny. A version of the tree with untransformed branches and including the accession numbers can be found in Figure S2.
The conserved motifs of each BnaSNARE protein sequence were identified by MEME and analyzed with the InterProScan tool (Figure 2b and Table S6). In brief, proteins in the same subclade seemed to share a similar motif composition, corresponding to the phylogenetic classification of BnaSNARE proteins. Motifs one and two correspond to the SNARE domains found in both Q- and R- SNARE proteins. Motifs six, seven, and eight, were found to be related to the syntaxin domains present in Q-SNAREs of B. napus, while motifs three, four, five, sixteen, and eighteen were found to be related to the Synaptobrevin and Longin domains present in R-SNAREs. In addition, motif fifteen is specific to Qbc SNARE, while motif nineteen is just present in BnaSYP3s. Along with the conserved motifs, the distribution of introns and exons in the 237 BnaSNAREs was analyzed with GSDS 2.0 (Figure 2c). We found a conserved number of introns within the subclades which is consistent with the phylogenetic classification. In detail, Qa-SNAREs contain various introns between 0 and 11. Among them, BnaSYP11s and BnaSYP12s contain the minimal introns 0 or 1. BnaSYP13s contain the most variable number of introns which are from 3 to 11. Almost all of the BnaSYP2s had six introns with two exceptions: BnaSYP22e and BnaSYP27 having eight and three introns, respectively. A similar situation also occurred in other types of BnaSNAREs (Figure 2c). Qb-SNAREs contain various introns between 1 and 9; Qc-SNAREs contain 3 to 11 introns; Qbc-SNAREs contain 3, 4, or 6 introns while R-SNAREs contain 1 to 23 introns. More so, R-SNAREs, BnaTYN11a, BnaTYN11b, BnaTYN11c, and BnaTYN11d harbor 23 introns each, which is the largest number detected in all the SNAREs in B. napus.
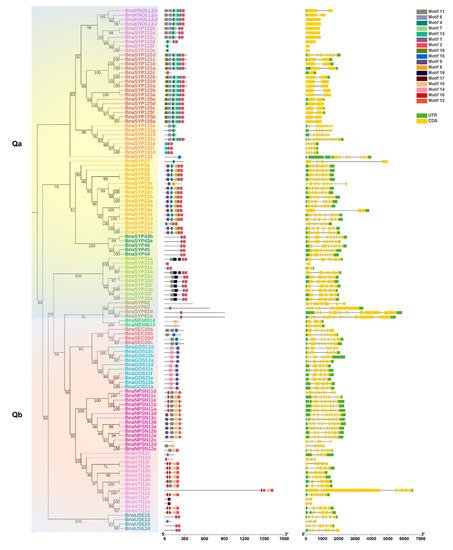
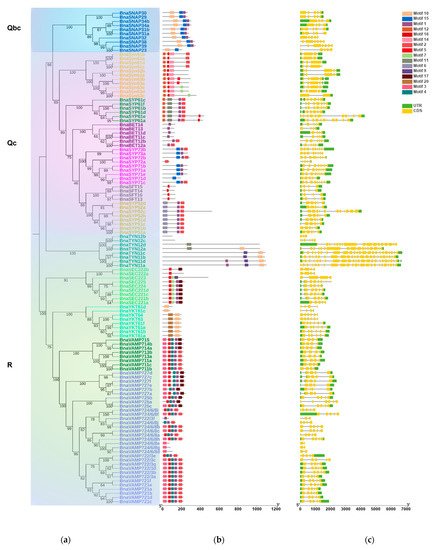
Figure 2. Gene intron/exon structures and protein conserved motifs of BnaSNAREs. (a) Phylogenetic tree of BnaSNARE proteins. (b) Conserved motif arrangements of BnaSNAREs. Twenty conserved motifs labeled with different colors were found in the BnaSNAREs sequences using the MEME program. Sequences of the conserved motif are presented in Table S5. (c) Exon-intron organizations of BnaSNAREs. The green boxes represent 5′or 3′ untranslated regions, yellow boxes represent exons, and black lines represent the introns. The lengths of the exons and introns can be determined by the scale at the bottom.
References
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. https://doi.org/10.1111/tpj.13669.
- Bayer, P.E.; Hurgobin, B.; Golicz, A.A.; Chan, C.-K.K.; Yuan, Y.; Lee, H.; Renton, M.; Meng, J.; Li, R.; Long, Y.; et al. Assembly and comparison of two closely related Brassica napus genomes. Plant Biotechnol. J. 2017, 15, 1602–1610. https://doi.org/10.1111/pbi.12742.
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005, 8, 135–141. https://doi.org/10.1016/j.pbi.2005.01.001.
- Cui, L.Y.; Wall, P.K.; Leebens-Mack, J.H.; Lindsay, B.G.; Soltis, D.E.; Doyle, J.J.; Soltis, P.S.; Carlson, J.E.; Arumuganathan, K.; Barakat, A.; et al. Widespread genome duplications throughout the history of flowering plants. Genome Res. 2006, 16, 738–749. https://doi.org/10.1101/gr.4825606.
- Grassi, A. de; Lanave, C.; Saccone, C. Genome duplication and gene-family evolution: The case of three OXPHOS gene fami-lies. Gene 2008, 421, 1–6. https://doi.org/10.1016/j.gene.2008.05.011.
- Zhao, W.; Dong, S.; Ye, W.; Hua, C.; Meijer, H.J.G.; Dou, X.; Govers, F.; Wang, Y. Genome-wide identification of Phytophthora sojae SNARE genes and functional characterization of the conserved SNARE PsYKT6. Fungal Genet. Biol. 2011, 48, 241–251. https://doi.org/10.1016/j.fgb.2010.11.006.
- Cucu, B.; Degreif, D.; Bertl, A.; Thiel, G. Vesicle fusion and fission in plants and yeast. Cell Calcium 2017, 67, 40–45. https://doi.org/10.1016/j.ceca.2017.08.007.
- Bonifacino, J.S.; Glick, B.S. The mechanisms of vesicle budding and fusion. Cell 2004, 116, 153–166. https://doi.org/10.1016/s0092-8674(03)01079-1.
- Söllner, T.; Bennett, M.K.; Whiteheart, S.W.; Scheller, R.H.; Rothman, J.E. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 1993, 75, 409–418. https://doi.org/10.1016/0092-8674(93)90376-2.
- Fasshauer, D.; Sutton, R.B.; Brunger, A.T.; Jahn, R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. USA 1998, 95, 15781–15786. https://doi.org/10.1073/pnas.95.26.15781.
- Sanderfoot, A. Increases in the number of SNARE genes parallels the rise of multicellularity among the green plants. Plant Physiol. 2007, 144, 6–17. https://doi.org/10.1104/pp.106.092973.
- Salinas-Cornejo, J.; Madrid-Espinoza, J.; Ruiz-Lara, S. Identification and transcriptional analysis of SNARE vesicle fusion regulators in tomato (Solanum lycopersicum) during plant development and a comparative analysis of the response to salt stress with wild relatives. J. Plant Physiol. 2019, 242, 153018. https://doi.org/10.1016/j.jplph.2019.153018.
- Sansebastiano, G.D.; Piro, G. The SNARE proteins (in plants) beyond the Nobel Prize. J. Plant Biochem. Physiol. 2014, 2, e122. https://doi.org/10.4172/2329-9029.1000e122.
- Wang, G.; Long, D.; Yu, F.; Zhang, H.; Chen, C.; Wang, Y.; Ji, W. Genome-wide identification, evolution, and expression of the SNARE gene family in wheat resistance to powdery mildew. PeerJ 2021, 9, e10788. https://doi.org/10.7717/peerj.10788.
- Rosenbaum, E.E.; Vasiljevic, E.; Cleland, S.C.; Flores, C.; Colley, N.J. The Gos28 SNARE protein mediates intra-Golgi transport of rhodopsin and is required for photoreceptor survival. J. Biol. Chem. 2014, 289, 32392–32409. https://doi.org/10.1074/jbc.M114.585166.
- Kienle, N.; Kloepper, T.H.; Fasshauer, D. Differences in the SNARE evolution of fungi and metazoa. Biochem. Soc. Trans. 2009, 37, 787–791. https://doi.org/10.1042/BST0370787.
- Burri, L.; Lithgow, T. A complete set of SNAREs in yeast. Traffic 2004, 5, 45–52. https://doi.org/10.1046/j.1600-0854.2003.00151.x.
- Furukawa, N.; Mima, J. Multiple and distinct strategies of yeast SNAREs to confer the specificity of membrane fusion. Sci. Rep. 2014, 4, 4277. https://doi.org/10.1038/srep04277.
- Kim, S. J.; Brandizzi, F. News and views into the SNARE complexity in Arabidopsis. Front. Plant Sci. 2012, 3, 28. https://doi.org/10.3389/fpls.2012.00028.
- Ben, Z.; Karnik, R.; YiZhou, W.; Wallmeroth, N.; Blatt, M.R.; Grefen, C. The Arabidopsis R-SNARE VAMP721 interacts with KAT1 and KC1 K+ channels to moderate K+ current at the plasma membrane. Plant Cell 2015, 27, 1697–1717. https://doi.org/10.1105/tpc.15.00305.
- Uemura, T.; Kim, H.; Saito, C.; Ebine, K.; Ueda, T.; Schulze-Lefert, P.; Nakano, A. Qa-SNAREs localized to the trans-Golgi network regulate multiple transport pathways and extracellular disease resistance in plants. Proc. Natl. Acad. Sci. USA 2012, 109, 1784–1789. https://doi.org/10.1073/pnas.1115146109.
- Chung, K.P.; Zeng, Y.; Li, Y.; Ji, C.; Xia, Y.; Jiang, L. Signal motif-dependent ER export of the Qc-SNARE BET12 interacts with MEMB12 and affects PR1 trafficking in Arabidopsis. J. Cell Sci. 2018, 131, jcs202838. https://doi.org/10.1242/jcs.202838.
- Lauber, M.H.; Waizenegger, I.; Steinmann, T.; Schwarz, H.; Mayer, U.; Hwang, I.; Lukowitz, W.; Jürgens, G. The Arabidopsis KNOLLE protein is a cytokinesis-specific syntaxin. J. Cell Biol. 1997, 139, 1485–1493. https://doi.org/10.1083/jcb.139.6.1485.
- Slane, D.; Reichardt, I.; El Kasmi, F.; Bayer, M.; Jürgens, G. Evolutionarily diverse SYP1 Qa-SNAREs jointly sustain pollen tube growth in Arabidopsis. Plant J. 2017, 92, 375–385. https://doi.org/10.1111/tpj.13659.
- Lian, Q.; Meng, Y.; Zhao, X.; Xu, Y.; Wang, Y.; Day, B.; Ma, Q. ShNPSN11, a vesicle-transport-related gene, confers disease resistance in tomato to Oidium neolycopersici. Biochem. J. 2020, 477, 3851–3866. https://doi.org/10.1042/BCJ20190776.
- Nühse, T.S.; Boller, T.; Peck, S.C. A plasma membrane syntaxin is phosphorylated in response to the bacterial elicitor flagel-lin. J. Biol. Chem. 2003, 278, 45248–45254. https://doi.org/10.1074/jbc.M307443200.
- Assaad, F.F.; Qiu, J.-L.; Youngs, H.; Ehrhardt, D.; Zimmerli, L.; Kalde, M.; Wanner, G.; Peck, S.C.; Edwards, H.; Ramonell, K.; et al. The PEN1 syntaxin defines a novel cellular compartment upon fungal attack and is required for the timely assembly of papillae. Mol. Biol. Cell 2004, 15, 5118–5129. https://doi.org/10.1091/mbc.e04-02-0140.
- Zhang, Z.; Feechan, A.; Pedersen, C.; Newman, M.-A.; Qiu, J.-L.; Olesen, K.L.; Thordal-Christensen, H. A SNARE-protein has opposing functions in penetration resistance and defence signalling pathways. Plant J. 2007, 49, 302–312. https://doi.org/10.1111/j.1365-313X.2006.02961.x.
- Catalano, C.M.; Czymmek, K.J.; Gann, J.G.; Sherrier, D.J. Medicago truncatula syntaxin SYP132 defines the symbiosome membrane and infection droplet membrane in root nodules. Planta 2007, 225, 541–550. https://doi.org/10.1007/s00425-006-0369-y.
- Kalde, M.; Nühse, T.S.; Findlay, K.; Peck, S.C. The syntaxin SYP132 contributes to plant resistance against bacteria and secre-tion of pathogenesis-related protein 1. Proc. Natl. Acad. Sci. USA 2007, 104, 11850–11855. https://doi.org/10.1073/pnas.0701083104.
- Zhu, J.; Gong, Z.; Zhang, C.; Song, C.-P.; Damsz, B.; Inan, G.; Koiwa, H.; Zhu, J.-K.; Hasegawa, P.M.; Bressan, R.A. OSM1/SYP61: A syntaxin protein in Arabidopsis controls abscisic acid-mediated and non-abscisic acid-mediated responses to abiotic stress. Plant Cell 2002, 14, 3009–3028. https://doi.org/10.1105/tpc.006981.
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. https://doi.org/10.1126/science.1253435.
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. https://doi.org/10.1111/tpj.13669.
- Bayer, P.E.; Hurgobin, B.; Golicz, A.A.; Chan, C.-K.K.; Yuan, Y.; Lee, H.; Renton, M.; Meng, J.; Li, R.; Long, Y.; et al. Assembly and comparison of two closely related Brassica napus genomes. Plant Biotechnol. J. 2017, 15, 1602–1610. https://doi.org/10.1111/pbi.12742.
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005, 8, 135–141. https://doi.org/10.1016/j.pbi.2005.01.001.
- Cui, L.Y.; Wall, P.K.; Leebens-Mack, J.H.; Lindsay, B.G.; Soltis, D.E.; Doyle, J.J.; Soltis, P.S.; Carlson, J.E.; Arumuganathan, K.; Barakat, A.; et al. Widespread genome duplications throughout the history of flowering plants. Genome Res. 2006, 16, 738–749. https://doi.org/10.1101/gr.4825606.
- Grassi, A. de; Lanave, C.; Saccone, C. Genome duplication and gene-family evolution: The case of three OXPHOS gene fami-lies. Gene 2008, 421, 1–6. https://doi.org/10.1016/j.gene.2008.05.011.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
05 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No