SNAREs (soluble N-ethylmaleimide-sensitive factor attachment protein receptors) are central components that drive membrane fusion events during exocytosis and endocytosis and play important roles in the different biological processes of plants. In this study, we identified 237 genes encoding SNARE family proteins in B. napus in silico at the whole-genome level. Phylogenetic analysis showed that BnaSNAREs could be classified into five groups (Q (a-, b-, c-, bc-) and R) like other plant SNAREs and clustered into twenty-five subclades. The gene structure and protein domain of each subclade were found to be highly conserved. In many subclades, BnaSNAREs are significantly expanded compared with the orthologous genes in Arabidopsis thaliana. BnaSNARE genes are expressed differentially in the leaves and roots of B. napus. RNA-seq data and RT-qPCR proved that some of the BnaSNAREs are involved in the plant response to S. sclerotiorum infection as well as treatments with toxin oxalic acid (OA) (a virulence factor often secreted by S. sclerotiorum) or abscisic acid (ABA), methyl jasmonate (MeJA), and salicylic acid (SA), which individually promote resistance to S. sclerotiorum. Moreover, the interacted proteins of BnaSNAREs contain some defense response-related proteins, which increases the evidence that BnaSNAREs are involved in plant immunity. We also found the co-expression of BnaSYP121/2s, BnaSNAPs, and BnaVAMP722/3s in B. napus due to S. sclerotiorum infection as well as the probable interaction among them.
- Brassica napus
- SNARE
- gene family
- Sclerotinia sclerotiorum
- expression profile
- hormone stimuli
1. Introduction
2.1 Identification of SNARE Genes in B. napus
2 Identification of SNARE Genes in B. napus
To identify all members of the SNARE family in B. napus, three methods including Pfam analysis, conserved domains search, and orthologous sequence BLAST were used. A total of 237 BnaSNAREs were identified. Of these genes, six (6) couples sharing 100% identity on the amino acid level but different nucleotide sequences were considered to be different BnaSNAREs. All candidate BnaSNAREs were named according to their best hit in Arabidopsis. Each gene name starts with an abbreviation for the species name B. napus (Bna), followed by the name of the most prominent Arabidopsis gene from this subclade (e.g., “BnaSYP122” for AtSYP122-like genes, “BnaSEC20” for AtSEC20-like genes, “BnaYKT61” for AtYKT61-like genes). Exceptions are BnaSNAP33s, BnaSNAP30s, and BnaSNAP29s which are named according to their protein molecular weight (e.g., “BnaSNAP31” represents the protein molecular weight is approximate 31 kDa). Genes on different chromosomes belonging to the same subclade were consecutively numbered according to their chromosome number from low-to-high values (e.g., four KNOLLE-like genes BnaC05g06210D, BnaA06g04950D, and BnaA08g26870D, BnaC08g13620D were named separately as “BnaKNOLLEa”, “BnaKNOLLEb”, “BnaKNOLLEc”, and “BnaKNOLLEd”). In the case of SYP4s, our phylogenies did not provide clear orthologous relationships among SYP41s, SYP42s, and SYP43s genes from B. napus and Arabidopsis. We therefore named the SYP4 subclade genes of B. napus as BnaSYP44, BnaSYP45, and BnaSYP46, taking up the current code of Arabidopsis. A similar strategy was adopted to name USE1, SFT1, MEMB1, and YKT62-like genes. VAMP724, VAMP726, and VAMP728 genes were similar to the case of BnaSYP4s, but the strategy did not fit this case because the codes in Arabidopsis are up to eight (VAMP728). “Slash” was rather used in naming these genes (e.g., “BnaVAMP724/6/8a”, “BnaVAMP724/6/8b”). Almost all of the identified SNAREs in B. napus showed the same conserved domain with their respective orthologs in Arabidopsis according to NCBI batch CD search, except for the 11 SYP6-like proteins, which just contain 1 N-terminal syntaxin-6 (PF09177) domain but lost a C-terminal SNARE domain. Furthermore, of these 11 SYP6-like proteins from B. napus, 3 orthologs from Arabidopsis were not previously identified. Considering the fact that the conserved N-terminal syntaxin-6 domain is unique to the SNARE family, we believe that the 11 genes belong to the SNARE family and we classified them as SYN-sub-family. The BnaSNARE proteins have varying physicochemical characteristics. Isoelectric points (pIs) of the proteins are between 4.44 and 11.84, and their molecular weights (MWs) range from 9.02 to 120.51 kDa. Exceptions are BnaSYP31d with 7.15 kDa, and both BnaSYP112e and BnaSYP112f have MW of 8.05 kDa, which fall below the range; BnaVTI11e, however, has an MW of 184.07 kDa which falls above the stated range. Subcellular localization prediction for the BnaSNARE proteins indicated that they are localized at the plasma membrane, ER, Golgi, vacuole, and a small group were located in the cytoplasm, mitochondrion, and nucleus.2.2 BnaSNAREs Belong to Well-Defined Subfamilies That Were Correlated to Their Gene Structures and Conserved Motifs
3 BnaSNAREs Belong to Well-Defined Subfamilies That Were Correlated to Their Gene Structures and Conserved Motifs
A maximum-likelihood phylogenetic tree of all the SNARE genes from A. thaliana and B. napus shows that the B. napus genome retains all the orthologs of A. thaliana SNAREs and the gene phylogeny roughly followed species phylogeny. In several subclades, one SNARE in A. thaliana is closely related to a double of two B. napus homologs (e.g., SFT1, USE1 Figure 1 and Table S4) which is consistent with the chromosome multiples of B. napus and A. thaliana as B. napus is heterotetraploid while A. thaliana is diploid. In many subclades, SNARE homologs in B. napus are significantly expanded compared to those in A. thaliana. Measured from the total point of view, the number of SNAREs in B. napus is much more than double of those in A. thaliana, in fact, nearly four times (e.g., TYN1, SEC20, SYP5 subclades Figure 1 and Table S1). The topology in BnaSYP12, BnaYKT6, BnaSYP6, and BnaVAMP72 subclades (Figure 1 and Table S1) is more complex, suggesting multiple duplication events, before and/or after polyploidization of B. napus. The BnaSNARE proteins displayed the same five groups described previously (Q (a-, b-, c-, bc-) and R) in A. thaliana and presented a similar proportion of members compared to A. thaliana (Table S5). Therefore, Qa-, Qb-, and Qc-BnaSNAREs are composed of 69, 44, and 37 (+11) genes, respectively. Qbc-BnaSNAREs have 10 members and R-BnaSNAREs have 65 members.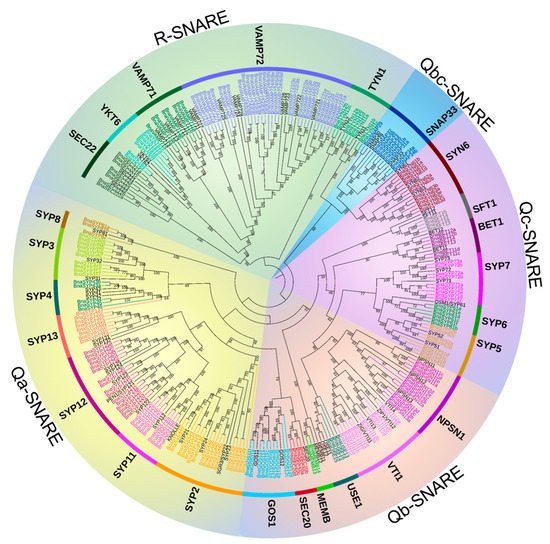
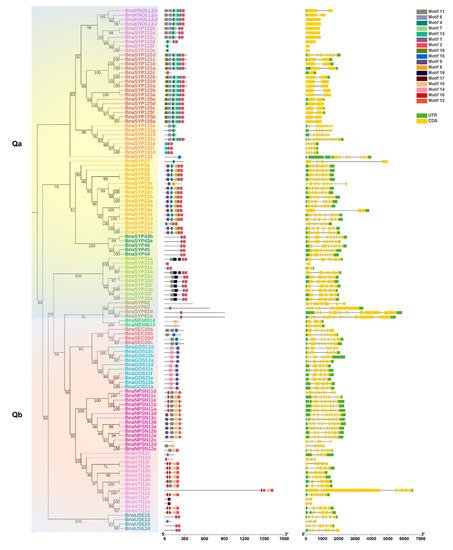
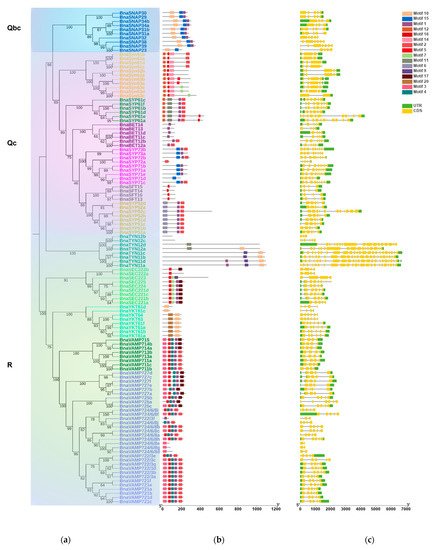
2.3. BnaSNAREs Exhibit a High Rate of Homolog Retention and Gene Duplication in the Genome
In many land plants, the number of identified SNAREs is between 50 and 70 [11,14]. Rice and Arabidopsis, which are both diploid, have a similar number of SNARE genes (60 and 64, respectively), while 173 SNAREs were found in wheat. The number of SNAREs in wheat is expected to be high considering the fact that wheat is an allohexaploid plant (60*3 = 180) (Table S5). B. napus contained the highest number of SNAREs in characterized land plant species and algae. Fifteen out of a total of twenty-five subclades contain significantly more than two times the number in rice and Arabidopsis (Figure 3, t-test, p < 0.001) and approach four times on the whole level (t-test, p = 0.11, 0.62).
|
|
|||
|
(a) |
(b) |
(c) |
(d) |
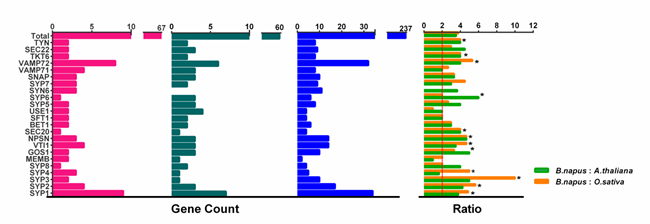
Figure 3. Number of SNARE genes. (a–c). The number of SNARE genes identified per SNARE-type subgroup in (a) A. thaliana; (b) O. sativa, and (c) B. napus []. (d). The ratio of total SNARE gene numbers to those in all subgroups is shown for B. napus: O. sativa (green) and B. napus: A. thaliana (orange). The expected ratio (2:1) in (d) is indicated by a red vertical line, and asterisks mark a significant deviation from the expected value (χ2 test, p < 0.05).
To better understand why SNAREs are so abundant in the B. napus genome, we analyzed in detail the homologous pairs from the A and C sub-genomes (Table 1). More than three-quarters (76.8%) of the 237 BnaSNARE genes identified are present in double (even as high as 89.7% when the “not categorized” gene is removed) (Tables 1 and S4).
Table 1. Homologous gene pairs of SNAREs in B. napus.
|
Homoeologous Group (A:C) |
All Genes in B. napus 1 |
BnaSNAREs (All) 2 |
BnaSNAREs (Omit Not Categorized) |
||||
|
Number of Gene Pairs |
Number of Genes |
% of Genes |
Number of Gene Pairs |
Number of Genes |
% of Genes |
||
|
1:1 |
80.8 |
91 |
182 |
76.8% |
91 |
182 |
89.7% |
|
0:1 |
6.75 |
7 |
7 |
3.0% |
7 |
7 |
3.4% |
|
1:0 |
12.4 |
11 |
11 |
4.6% |
11 |
11 |
5.4% |
|
Other ratios 3 |
- |
3 |
3 |
1.3% |
3 |
3 |
1.5% |
|
Not categorized |
- |
- |
34 |
|
- |
- |
|
|
Total |
|
|
237 |
100.0% |
|
203 |
100.0% |
1 [32]. 2 See Table S4. 3 Other ratios: one of the homologous not belong to SNAREs. 4 Not categorized: genes located on random chromosomes.
We mapped the 237 BnaSNAREs on 10 An and 9 Cn chromosomes and found that they are unevenly distributed in B. napus. A total of 121 BnaSNARE genes were mapped on the An sub-genome, while 116 were mapped on the Cn sub-genome (Figure 4 and Table S1). Chromosomes A03 (17) and C03 (19) contain the highest number of BnaSNAREs in the An and Cn sub-genomes, respectively. The lowest number of BnaSNAREs was found on chromosomes A02 and C01 where each contains six. The remaining chromosomes harbor between 7 and 18 BnaSNAREs. Additionally, 11 and 8 members of the BnaSNAREs from A and C sub-genomes, respectively, could not be mapped to a particular chromosome. Based on their chromosome positions, we identified 22 clusters of tandem duplication cases, including 47 BnaSNARE genes (47/237, 19.8%) (Table S7), suggesting a role of tandem duplication events in the expansion of BnaSNARE gene members.
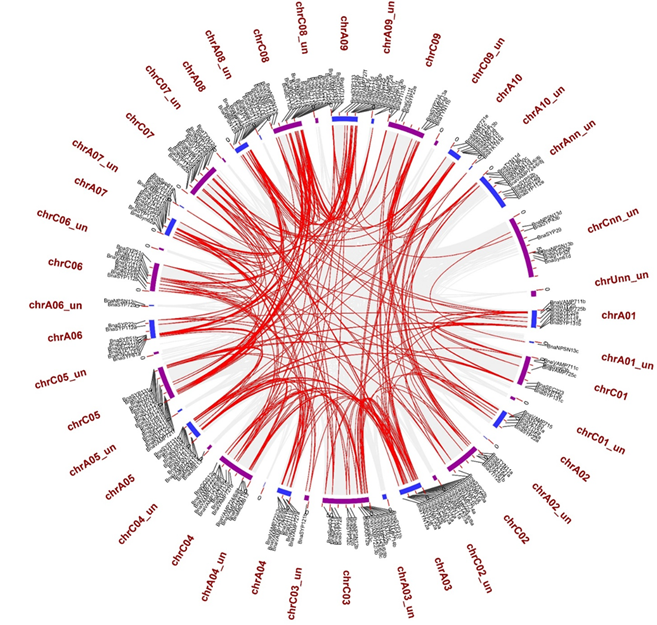
Figure 4. The locations on chromosomes and homologous relationships of the BnaSNAREs. All BnaSNAREs were mapped to their respective locus in the B. napus genome in a circular diagram using Advanced Circos of TBtools. An and Cn sub-genomes are indicated by shades of blue and purple colors.
The duplication analysis showed explosive gene duplication events (288 duplication events including 206 BnaSNARE genes) in BnaSNAREs (Table S8). The sub-family BnaVAMP72s have the largest number of duplication events (55/288, 19.1%) in the B. napus genome, followed by BnaSYP1s (35/288, 12.2%) (Figure 4 and Table S8). To determine the possible selection constraints on the duplicated BnaSNAREs, we estimated the Ka/Ks ratio for each pair of paralog genes (Table S8). We found that the Ka/Ks ratios of the duplicated BnaSNAREs gene pairs are all less than 1. Besides, the Ka/Ks value of 97.9% (282/288) duplicated gene pairs is less than 0.5, suggesting a high level of purifying selection stress of evolution.
2.4. Differential Expression of BnaSNAREs in Leaves and Roots of B. napus
To understand the biological functions of BnaSNAREs, we first detected the expression profiles of BnaSNARE genes in leaves and roots of B. napus (Table S9 and Figure S2). The RPKM values of BnaSNAREs were downloaded from genoscope (http://www.genoscope.cns.fr/brassicanapus/, accessed on 26 October 2020). BnaSNAREs were expressed differentially in the roots and leaves of B. napus. Most of the root-expressed genes were expressed unequally in leaves (e.g., BnaSNAPs and BnaSYP12s were highly expressed in roots but not in leaves). In total, expression levels of more than half of the BnaSNARE genes were higher in root than in leaves. The RPKM of 37 BnaSNAREs was drastically low and negligible expression was recorded in roots, leaves, or neither. The BnaSYP1s contributed the most low-expressed genes followed by the BnaVAMP7s. The reason for this low expression was probably due to tissue specificity in the expression or the presence of processed pseudogenes.
2.5. BnaSNAREs Are Involved in Regulation of B. napus Resistance to S. sclerotiorum
S.sclerotiorum is one of the main pathogens causing serious stem rot disease of B. napus [38]. To improve our understanding of the role of BnaSNAREs during the infection of S. sclerotiorum, we inoculated a resistant variety Zhongshuang9 and a susceptible variety 84039 and collected samples at 0 hpi, 12 hpi, and 22 hpi, then performed RNA extraction, cDNA library construction, and sequencing. The RNA-seq data showed that the biotic stress caused by S. sclerotiorum resulted in the accumulation of BnaSNARE transcripts (Figure 5 and Table S10). The expression levels of the well-known resistance-related Qa-SNARE SYP121(pen1) orthologs, BnaSYP121s and BnaSYP122s, were highly up-regulated both in varieties 84039 and Zhongshuang9 at 12 h and even higher at 22 h after inoculation with S. sclerotiorum. Furthermore, other genes belonging to BnaSYP21s, BnaSYP32s, BnaSYP52s, BnaSNAP33s, BnaGOS11s, BnaVTI12s, BnaVAMP7s, and BnaSYN6s were also up-regulated after inoculation with S. sclerotiorum. Among them, BnaSYP21b, BnaSYP21c, BnaSYP32b, BnaSYP61d, BnaSYP63d, BnaSNAP31a, BnaSNAP31b, BnaSNAP34a, BnaSNAP34b, and BnaVAMP722/3s were up-regulated in both 84039 and Zhongshuang9 varieties, while BnaSYP22b, BnaSYP32d, BnaMEMB13, BnaGOS11b, and BnaUSE14 were mainly up-regulated in the resistant variety Zhongshuang9 and some other genes as BnaSYP43a and BnaSYN61c were up-regulated in the susceptible variety 84039. Some BnaSNARE genes were down-regulated in response to inoculation with S. sclerotiorum, such as BnaKONLLEa, BnaSYP83, BnaVTI11f, BnaNPSN12b, BnaNPSN12e, and BnaSYN62s. Almost all members of BnaSYN61s and BnaSYN63s genes showed a trend becoming up-regulated in both 84039 and Zhongshuang9 varieties in response to S. sclerotiorum, while BnaSYN62s were down-regulated, implying that BnaSYN6s are associated with plant resistance.
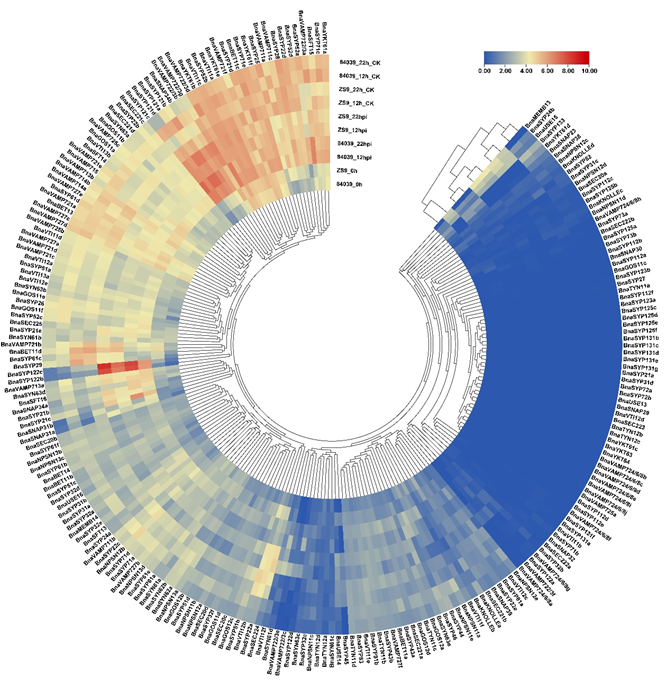
Figure 5. Expression of BnaSNAREs in response to the infection of S. sclerotiorum. The transcription level of BnaSNAREs in leaves at 12 h and 22 h post-inoculation with S. sclerotiorum is shown as a heatmap. Data were normalized by log2 (FPKM+1). The cluster tree of the BnaSNARE genes based on the expression level is shown at the center and top.
To further verify the above results, we performed real-time quantitative reverse transcription-PCR (RT-qPCR) to detect the expression level of BnaSYP1s (Figure 6). Sixteen of the tested BnaSYP1s showed either induction or suppression in response to S. sclerotiorum infection, while others had too weak transcript abundances to detect. The expression levels of BnaKNOLLEc and BnaKNOLLEd were decreased at all time points and reached the lowest level at 24 hours. On the contrary, BnaSYP121s and BnaSYP122s were highly up-regulated. The expression of BnaSYP125s changed slightly at each time point in variety 84039 but these changes were highly pronounced in the Zhongshuang9 variety. Several genes belonging to one subclade showed opposite expression level trends: BnaSYP123a was up-regulated while BnaSYP123b was down-regulated in both varieties; BnaSYP131a was up-regulated in variety 84039 but down-regulated in variety Zhongshuang9 while the reverse was the case for BnaSYP131d gene expression.
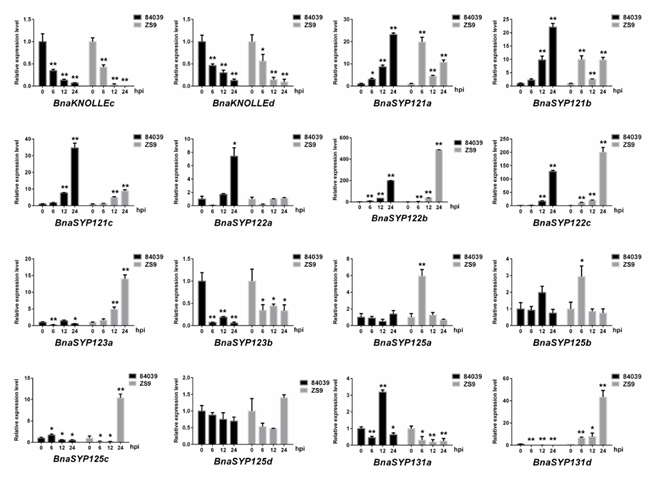
Figure 6. Expression of BnaSYP1-type genes in resistant variety Zhongshuang9 and susceptible variety 84039 of B. napus at different times after inoculation with S. sclerotiorum. RT-qPCR data were calculated by the methods of 2-ΔΔCT. STDEV is indicated as error bars. All data are the mean of 3 biological replicates. Significant differences are shown by * (p < 0.05) and** (p < 0.01).
2.6. BnaSYP1s Respond to Phytohormones and Oxalic Acid Treatments
SNAREs were found to participate in plant development [1,3,29], abiotic stress [12,31], and pathogen resistance pathways [21,25,30,39–41]. Furthermore, MeJA, SA, and ABA were proved to regulate plant resistance to S. sclerotiorum [42–46]. B. napus varieties Zhongshuang9 and 84039 were treated with exogenous OA, MeJA, SA, and ABA. RT-qPCR results showed that most of the tested genes from BnaSYP1, BnaSYP2, BnaSYP3, and BnaSYP52 were sensitive to all treatments (Figure 7 and Table S11). Most of the tested genes showed notably increased expression levels to a large degree in B. napus variety Zhongshuang9 after treatment with ABA, while several genes were up-regulated to more than 2-fold in variety 84039. After treatment with MeJA, transcripts from many genes could not be detected. A few genes such as BnaSYP112c, BnaSYP121d, BnaSYP122b, BnaSYP21c, BnaSYP32c, and BnaSYP32e were induced to a larger degree in the resistant variety than in the susceptible variety; while BnaKNOLLEs was significantly down-regulated in the resistant variety compared to the susceptible variety. BnaSYP121c, however, was significantly down-regulated in the susceptible variety in comparison to the resistant variety. The SA-induced transcript changes of BnaKNOLLEs, BnaSYP123a, BnaSYP125b, BnaSYP125c, BnaSYP131a, and BnaSYP131c were increased in both resistant and susceptible varieties, while BnaSYP122a, BnaSYP122d, BnaSYP21e, BnaSYP29, BnaSYP32,c and BnaSYP52b were suppressed by SA in both varieties. The expression of BnaSYP122b was induced in the susceptible variety but inhibited in the resistant variety whereas BnaSYP25 showed the opposite trend. BnaKNOLLEs, BnaSYP121a, BnaSYP121b, BnaSYP121d, BnaSYP122d, BnaSYP24a, and BnaSYP29 showed similar expression pattern as those induced by MeJA.
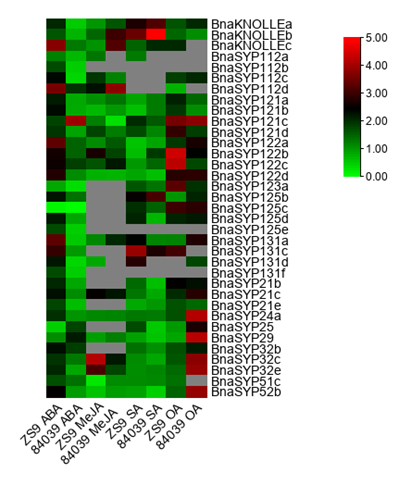
Figure 7. Expression profiles of Qa-BnaSNAREs in resistant variety Zhongshuang9 and susceptible variety 84039 of B. napus under treatment with ABA, MeJA, SA, and OA. Green and red colors are used to represent low-to-high expression levels, and colors scales correspond to the fold-change values compared with the counterpart control. RT-qPCR data were calculated by the method of 2-ΔΔCT and appeared after Log2 conversion. The gray color represents no available data. All values were detailed in Table S11.
OA treatments also induced the different accumulation of BnaSNAREs transcripts. Most of the tested BnaSNAREs were up-regulated by more than two-fold under treatment with OA, especially in the susceptible variety 84039. The expression levels of BnaSYP24a, BnaSYP25, BnaSYP29, BnaSYP32c, BnaSYP32e, and BnaSYP52b increased to a very high level in variety 84039, whereas those of other genes such as BnaSYP121d, BnaSYP122b, BnaSYP122c, and BnaSYP123a were highly induced in the resistant variety Zhongshuang9. Besides, transcripts of BnaKNOLLEa, BnaSYP112c, BnaSYP121c, BnaSYP122d, BnaSYP125c, BnaSYP125d, BnaSYP21b, BnaSYP21e, and BnaSYP32b showed similar trends in both 84039 and zhongshuang9 varieties under OA treatment. These results suggest that BnaSNAREs participate in diverse signaling pathways through complicated functional mechanisms.
2.7. BnaSYP121/2s Expressions Highly Correlate with Those of BnaSNAPs and BnaVAMP722/3s during S. sclerotiorum Infection
The above results showed that BnaSYP121/2s was induced by ABA, MeJA, SA, and OA treatments as well as S. sclerotiorum infection, indicating a significant role of BnaSYP121/2s in pathogen resistance. SNARE proteins usually form complexes and play important biological functions in the form of complexes [47]. In addition, it has been reported that SYP121 interacts with the SNARE proteins SNAP33, VAMP721, and VAMP722 to participate in the process of powdery mildew resistance. The correlations of expression patterns among BnaSYP121s, BnaSYP122s, BnaSNAPs, BnaVAMP721s, and BnaVAMP722/3s during S. sclerotiorum infection were analyzed in this paper (Figure 8a). The expression patterns of several genes such as BnaSNAP31a, BnaSNAP31b, BnaSNAP34a, BnaSNAP34b, and BnaVAMP722/3s showed positive correlations (r ≥ 0.6) with BnaSYP121/2s, while BnaSNAP32 and BnaSNAP38 showed negative correlations with BnaSYP122s (r ≤ −0.6). Several pairs of members among BnaSYP121s and BnaSYP122s showed a high level of correlation (r ≥ 0.87), which implies the possibility of functional redundancy.
|
|
|
|
(a) |
(b) |
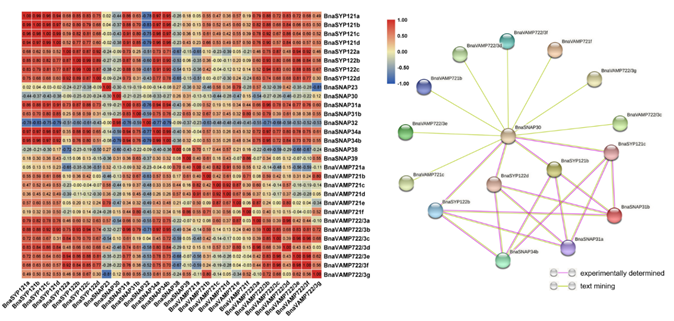
Figure 8. Correlations between the gene expression patterns of BnsSYP121/2s, BnaSNAPs, and BnaVAMP721/2/3s (a) and putative interaction networks among these proteins (b).
To further uncover the potential association of BnaSYP121/2s, BnaSNAPs, and BnaVAMP722/3s, protein–protein interaction analysis was conducted using STRING-DB (Table S12). Except for 15 undetected proteins, the association among the other 16 proteins is shown in Figure 8b. According to STRING-DB, the interaction patterns of BnaSYP121s and BnaSYP122s with other proteins were exactly the same, and they all interacted with BnaSNAP30, BnaSNAP31a, BnaSNAP31b, and BnaSNAP34b. While BnaVAMP721s and BnaVAMP722/3s only interact with SNAP30, we speculate that SNAP30 may be the key factor in the formation of the SNARE complex. These results showed the co-expression and potential interaction of BnaSYP121/2s, BnaSNAPs, and BnaVAMP722/3s under fungal pathogen infection, suggesting that BnaSYP121/2s may perform anti-fungal functions through the formation of BnaSYP121/2s-SNAPs-VAMP722/3s complexes.
2.8. BnaSNAREs Interaction Networks Indicate They Mainly Function in Vesicle-Mediated Transport, Protein Localization, and Response to Abiotic or Biotic Stress
To further investigate the interacted proteins of BnaSNAREs, we built interaction networks of AtSNAREs, and the corresponding orthologs in B. napus were identified. A total of 1574 Arabidopsis proteins were found to interact with AtSNAREs (Table S13). By syntenic analysis, 5184 syntenic orthologs were identified in B. napus (Table S14). The interaction networks of AtSNAREs showed a very complicated correlation with other proteins including not only the SNARE family members but also many other transport-related proteins, protein receptors, kinases, and so on, which indicate BnaSNAREs are involved in several mechanisms by regulating many downstream factors or being regulated by many upstream proteins.
Gene ontology (GO) enrichment analysis of proteins in BnaSNAREs interaction networks was conducted to reveal their functional characteristics. The most enriched and meaningful BP terms were related to vesicle-mediated transport and localization of proteins (Figure 9), illustrating the conserved functions of SNARE proteins in transporting and localizing proteins in plant cells. Meanwhile, BnaSNAREs were also related to some defense response signaling pathways, such as “defense response to oomycete”, “anion transmembrane transport”, “calcium ion transport”, and “organic acid transport”, indicating that SNARE protein may be involved in plant response to abiotic and biotic stress through association with these proteins. These results will shed light on their undetermined functions in other biological processes.
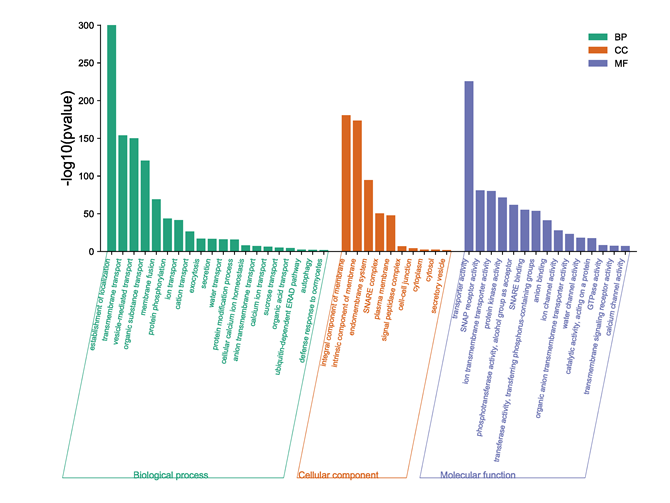
Figure 9. The gene ontology of genes in BnaSNAREs interaction networks.
- Materials and Methods
3.1. Identification of SNAREs in B. napus
To identify all candidate members of the SNARE family in B. napus, we combined three different methods. First, we identified the conserved domains and Pfams in all of the 64 SNAREs from A. thaliana (https://www.arabidopsis.org/, accessed on 23 August 2019) using the online batch CD-search server (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi, accessed on 25 August 2019 ). We generated 10 Pfams and 12 conserved domains specific for the SNARE superfamily (Table S2) as previously reported [12]. Then, genome-wide analysis of the Pfams was conducted and proteins containing the above Pfams were considered as SNARE protein candidates. Second, we searched for SNARE conserved domains and Pfams annotation in the B. napus genome annotation resources (http://www.genoscope.cns.fr/brassicanapus/, accessed on 26 October 2021) where proteins annotated with any of the 10 Pfams or the 12 conserved domains were identified as SNARE protein candidates. Third, the sequences of the 64 SNARE proteins in A. thaliana were used for similarity search against B. napus proteome by BLASTp. Finally, all possible BnaSNAREs identified by these three methods were validated by CDD (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi, accessed on 14 January 2020), Pfam (http://pfam.xfam.org/, accessed on 21 January 2020) and SMART (http://smart.embl-heidelberg.de/, accessed on 16 January 2020) analyses.
Biochemical parameters such as length of sequences, molecular weights, and isoelectric points of BnaSNAREs were calculated by the ProtParam tool (https://web.expasy.org/protparam/, accessed on 19 January 2020) and the subcellular localization was predicted by three different tools: Plant-mPLoc [48], CELLO [49], and pLoc-mEuk [50].
3.2. Phylogenetic Analysis
SNARE protein sequences from B. napus and A. thaliana were aligned using MAFFT [51,52]. Then, a phylogenetic tree was generated using IQ-server (http://iqtree.cibiv.univie.ac.at/ accesed on 22 November 2021)[53] as follows: the substitution models were calculated with ModelFinder (integrated with IQ-TREE; best-fit model: JTT+I+G4 chosen according to the Bayesian information criterion) [54]. Subsequently, a phylogenetic tree was generated using Ultrafast bootstraps as well as a Shi-modaira–Hasegawa approximate likelihood ratio test (SH-aLRT) test (1000 replicates each) [55–57]. The resulting tree file was visualized with iTOL v 5 [58] (https://itol.embl.de/, accessed on 28 November 2021).
3.3. Gene Structure and Protein Conserved Motifs
The gene structure information was displayed in the Gene Structure Display Server [59] (GSDS; http://gsds.cbi.pku.edu.cn, accessed on 05 March 2020). Conserved motifs of these genes were determined using the MEME suite [60] (http://meme-suite.org/tools/meme, accessed on12 March 2020) with the following parameters: optimum motif widths of 6–50 residues and a maximum of 50 motifs. Then, these motifs were searched using the InterProScan tool [61] (https://www.ebi.ac.uk/interpro/search/sequence-search, accessed on 15 March 2020). The schematic diagram of the amino acid motifs for each SNARE gene was drawn accordingly using Advanced Gene Structure View of TBtools [62].
3.4. Chromosomal Spread, Gene Duplication, and Collinear Analysis
The locations on the chromosomes of BnaSNAREs were shown by Circos [63]. Detection of putative gene duplication events was done with MCScanX (E-value 10−5) [64] and visualized using Advanced Circos of TBtools software v1.098 [62]. Tandem duplication events were defined as two or more homologous genes located on a chromosomal region within 200 kb [65].
3.5. Transcriptional Profile of BnaSNAREs in Leaves and Roots
Expression profiles of BnaSNAREs in leaves and roots of B. napus variety “Darmor-bzh” were downloaded from the B. napus genome database (https://www.genoscope.cns.fr/brassicanapus/, accessed on 26 May 2020). Reads per kilobase million (RPKM) values of all the BnaSNAREs were extracted and submitted to TBtools to generate heatmaps. All of the heatmaps were normalized by Log2 (value).
3.6. Plants and Fungal Materials and Growth Conditions
The susceptible (84039) and resistant (Zhongshuang9) varieties of B. napus [66] (Figure S3) were provided by Shengyi Liu of the Oil Crops Research Institute, Chinese Academy of Agricultural Sciences. Sclerotia of the fungus S. sclerotiorum 1980 were germinated to produce hyphal inoculum on potato dextrose agar (PDA) at 22 °C. The B. napus used in these experiments were grown in pots containing soil and vermiculite (3:1 v/v) under greenhouse conditions at 23–25 °C with a photoperiod of 16/8 h light/dark, fertilization with commercial N:K:P (1:1:1) every 10 d.
3.7. Transcriptional Profiling of BnaSNAREs during S. sclerotiorum Infection
After 4 weeks of growth, the 3rd/4th leaves of the B. napus were inoculated with agar plugs excised from the edges of growing S. sclerotiorum colonies; three biological replicates for the leaf samples were detached at 12 hpi and 22 hpi and sent to Novogene Co., Ltd., (Beijing, China) for RNA extraction, library construction, and transcriptome sequencing using Illumina sequencing platform. After removing the 5′ and 3′ -adapters, N> 10% sequences, low-quality sequences (sequence quality values <= Q20), the clean data were aligned to the B. napus reference genome (, accessed on 26 October 2020) using TopHat2 [67] (http://ccb.jhu.edu/software/tophat/index.shtml, accessed on 24 March 2016). The transcript abundance (in RPKM value) of each gene was calculated by HTSeq [68] (https://htseq.readthedocs.io/en/release_0.11.1/, accessed on 24 March 2016).
3.8. Plant Treatments, RNA Isolation, and RT-qPCR
The 3rd and 4th leaves of four-week-old Zhongshuang9 and 84039 were sprayed with MeJA, SA, ABA, and OA or inoculated with S. sclerotiorum strain 1980 and collected at 0, 6, 12, 24, and 48 hours post-inoculation (hpi) and immediately frozen in liquid nitrogen and stored at −80 °C. Three biological replicates for inoculated leaves were maintained.
The total RNA isolation and purification of the samples were performed using RNAprep Pure Plant Plus Kit (Polysaccharides and Polyphenolics-rich) (TIANGEN, Beijing, China). All the RNA isolations for gene expression were done in triplicate for each sample analyzed. RNA integrity was visualized by 1% agarose gel electrophoresis. Their concentrations and purities (OD260/OD280 ratio > 1.95) were determined with a NanoDrop One Microvolume UV-Vis Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Exactly 1 μg of total RNA were reversely transcribed in a 20 μL reaction mixture using PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, , China) following the manufacturer’s instructions to remove traces of contaminant DNA and prepare cDNA.
Quantitative real-time PCR (RT-qPCR) analysis was used to analyze the expression levels of the identified BnaSNAREs. The standard RT-qPCR with SYBR Premix Ex Taq II (TaKaRa, Beijing, China) was repeated at least three times on a CFX96 Real-Time System (BioRad). The results were analyzed using the 2−(∆∆Ct) method, using BnaACTIN as the endogenous reference gene [69]. The primers used in this study are listed in Table S15.
3.9. Protein–Protein Interaction Analysis and Gene Ontology Analysis
The STRING-DB (v 11.5, https://string-db.org/, accessed on 26 November 2021) online service was used to predict putative protein–protein interaction networks among candidate proteins of BnaSNAREs. The interaction network of AtSNAREs in A. thaliana was constructed using the Arabidopsis Interactions Viewer (http://bar.utoronto.ca/interactions/cgi-bin/arabidopsisinteractionsviewer.cgi, accessed on 26 November 2021). Interacted proteins of AtSNAREs were replaced with corresponding syntenic orthologs in B. napus using syntenic analysis mentioned above. The GO enrichment analysis of BnaSNAREs interacted proteins was conducted by g: GOSt of g: Profiler (https://biit.cs.ut.ee/gprofiler/gost, accessed on 09 December 2021).
- Discussion
The heterotetraploid nature of B. napus and the large size of the BnaSNARE family provide an ideal opportunity to study the evolutionary fates of these genes. The BnaSNARE family (with 237 members) is one of the largest families of proteins among flowering plants [11,12] and has c.3.5 times as many as Arabidopsis (237:67~3.5). Another well-analyzed protein family in B. napus is NBS-LRR, which has c. 2.9 times as many as Arabidopsis (425:149~2.9) [32,70], while fatty acid desaturase family and NAC transcription factor family were expanded to c.3.4 and c.3.6 times as many as in Arabidopsis, respectively [71].
In fact, a variety of duplication patterns is observed in some subclades of BnaSNAREs. First, many sequences of BnaSNAREs are truncated (e.g., BnaSYP122b, BnaNPSN12c, BnaSYP72a) (Table S1 and Figure 2b,c), indicating that gene amplification occurred through transposable elements. Second, 47 genes from the BnaSNAREs are found in close proximity to each other, pointing towards tandem duplications as a mechanism for family expansion (Table S7). Third, each of the two Arabidopsis SYP2 genes (AtSYP21 and AtSYP23) has five and six paralogous genes in B. napus (Table S1). The phylogenetic analysis and gene position on the chromosome of these genes suggests gene duplications in the lineage leading to Brassica but before the polyploidization of B. napus. It has been found that numerous regions that are homologous to the Arabidopsis genome were triplicated within the diploid species of Brassica [72,73]. The amphidiploid species B. napus has an A genome coming from B. rapa and a C genome coming from B. oleracea. Furthermore, genes are essentially conserved within these two genomes [73], and this presents a large proportion of segmental or WGD duplication events between An and Cn genomes. We speculate that the gene expansion of SNAREs in B. napus is a synergistic effect of polyploidization and hybridization working together.
We found that 90.3% of all the BnaSNARE genes were expressed in roots or leaves or both, and the expression was relatively low when compared with the genome-wide assessment [32]. BnaSYP1s and BnaVAMP7s occupied most of the low transcript abundance genes. Some of these genes may constitute pseudogenes. Because several members of SYP1s and VAMP7s in other plants were proved to be involved in resistance to biotrophic or semi-biotrophic pathogen infections [2,29,30,39,74], the expressions of BnaSYP1s and BnaVAMP7s might be induced by certain conditions, thereby making them good candidates for investigating variety-specific resistance to biotic stress. Whether these genes are still functional remains an open question. In some cases, pseudogenes may contribute to the regulation of gene expression and generating genetic diversity by, for example, providing transcription factors (TFs) and RNA polymerase II (Pol II) binding sites [75].
The expression level of genes belonging to the same sub-family is diverse, which may be related to the fact that different members of the same sub-family play diverse roles in the same life activities. In wheat, silencing TaNPSN11/13 but not TaNPSN12 reduced resistance to Puccinia striiformis f. sp. tritici (Pst) virulent race CYR2 [41]. In rice, both OsVAMP711 and OsVAMP714 belong to the VAMP71 sub-family; overexpression of OsVAMP714 enhanced resistance to rice blast while that of OsVAMP7111 does not [40]. VAMP721/722/724 but not VAMP711/727 are involved in SA-associated apoptosis by interacting with PVA31 to combat pathogen infection [76]. Though having different functions mediated by the same SNARE homologs, functional redundancy has also been reported for these genes. For example, AtSYP123, AtSYP125, and AtSYP131 of the SYP1 sub-family were necessary for the development of male gametophyte in Arabidopsis [24]. The VTI1 SNARE family consists of four genes (AtVTI11, AtVTI12, AtVTI13, and AtVTI14) in Arabidopsis but only AtVTI11 and AtVTI12 are expressed at significant levels [77–80].AtVTI11 and AtVTI12 have different functions in vesicle trafficking to the vacuole, while AtVTI11 and AtVTI12 can functionally substitute each other when forming a complex with AtSYP41 and AtSYP42 to drive vesicle fusion [80–82]. Functional overlap was also observed among AtSYP4 family members [82]. AtSYP121/122 form complex with AtSNAP33 and AtVAMP721/722 during defense against powdery mildew, but the complexes so formed with AtVAMP724 and AtVAMP727 are not related to plant immunity [2]. Our RNA-seq data showed similar trends: several members of BnaVAMP722/3s were induced by S. sclerotiorum infection, while most of the BnaVAMP727s members were unaffected.
Diseases caused by S. sclerotiorum (Lib.) de Bary is a serious threat to the production of B. napus [38]. S. sclerotiorum causes the rotting of leaves, stems, and pods of B. napus and results in a considerable loss of seed yield around the world [83]. The main factors that promote S. sclerotiorum pathogenicity and colonization are oxalic acid and cell-wall-degrading enzymes secreted by the fungus. Oxalic acid not only acts as a pathogenic factor but also detoxifies S. sclerotiorum in the later stages of infection [84]. Our RNA-seq data and RT-qPCR results showed consistency of up-regulated BnaSNAREs (including BnaKNOLLEs, BnaSYP121s, and BnaSYP122s) in response to S. sclerotiorum and OA treatment.
In recent years, many researchers began to explore the signal pathways involved in the interaction between S. sclerotiorum and B. napus or other plants. They found that the disease resistance signals induced by S. sclerotiorum are mainly mediated by JA and ABA [85], which are also affected by ethylene and SA signaling pathways [45]. Our data showed that most of the BnaSYP1-type genes are differently induced by SA, JA, and ABA, suggesting a potential role in the interactions between B. napus and S. sclerotiorum.
Previous studies have shown that SNARE proteins often perform biological functions in the form of complexes in cells [47]. For example, PEN1/SYP121 can form tetramers with one scaffold protein SNAP33 and two vesicle-related membrane proteins VAMP721 and VAMP722, which jointly regulate the fusion process of vesicles and plasma membrane, and ultimately mediate plant disease resistance to pathogens [74]. Because SYP122 and PEN1/SYP121 are homologous proteins, the Yoichiro Fukao research group has identified through interaction genomics that SYP122 interacts with PEN1/SYP121 [27,39]. We infer that SYP122 may also interact with the above four proteins (SYP121, SNAP33, VAMP721, and VAMP722). For this reason, we analyzed the expression levels of BnaSYP121s, BnaSYP122s, BnaSNAPs, BnaVAMP721, and BnaVAMP722/3s during S. sclerotiorum infection and found that the expression levels of the several genes were highly correlated. The interaction network analysis of these proteins further proved the association between BnaSYP121/122s and BnaSNAPs, BnaVAMP721, and BnaVAMP722/3s, so we speculate that BnaSYP121/122 may form a complex with BnaSNAPs and BnaVAMP722/3s to promote the resistance of plants against S. sclerotiorum. However, the functional mechanism of these SNARE proteins needs to be confirmed by further experiments.
- Conclusions
A total of 237 BnaSNAREs were identified, which is the highest number of the protein family unveiled in all species previously studied. The expansion of the BnaSNARE family is primarily due to polyploidization and hybridization events. Besides, RNA-seq data showed the expression of BnaSNAREs in leaves and roots of B. napus. The expression profiles under the influence of S. sclerotiorum implied the potential role of BnaSNAREs in mediating the resistance of B. napus against S. sclerotiorum. Differential expression trends were also observed following RT-qPCR under SA, MeJA, ABA, and OA treatments, signifying the important role of BnaSNAREs in multiple signaling pathways. Moreover, the interaction protein of BnaSNARE contains some defense response-related proteins, which increases the evidence that BnaSNARE protein is involved in plant immunity. Additionally, members of BnaSYP121/2s, BnaSNAPs, and BnaVAMP722/3 showed probable interaction and correlated expression profiles upon infection with S. sclerotiorum. Although elucidating the exact functional mechanism of these BnaSNAREs in biotic and abiotic conditions requires further analysis, our findings provide the first gene-family-wide survey on the expression patterns of specific B. napus SNAREs in pathological conditions, and these highly up- and down-regulated genes can serve as candidate genes for future investigations.
Supplementary Materials: The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: Maximum-likelihood phylogeny of SNARE proteins from B. napus and A. thaliana Branches not transformed; Figure S2: Expression profiles of BnaSNAREs in roots and leaves of B. napus; Figure S3: Lesions in the leaves of B. napus varieties Zhongshuang9 and 84039 at different times after inoculation with S. sclerotiorum; Table S1: The characteristics of SNARE genes from B. napus; Table S2: Conserved domains of SNAREs in Arabidopsis thaliana predicted by CDD-Batch; Table S3: Subcellular localizations of BnaSNARE proteins; Table S4: Homologous gene pairs of SNAREs among An and Cn sub-genomes of B. napus and their putative orthologs in A. thaliana; Table S5: Grouped number of SNARE proteins in Triticum aestivum, Oryza sativa, Solanum lycopersicum, Arabidopsis thaliana, and Brassica napus; Table S6: Twenty conserved motifs of BnaSNAREs defined by the MEME suite; Table S7: Tandem duplication events of BnaSNARE genes; Table S8: The estimated Ka/Ks ratio for each pair of paralog genes from B. napus; Table S9: RNA-Seq data of BnaSNAREs in root and leaf. Read abundance is shown in terms of reads per kilobase million; Table S10: RNA-seq expression of BnaSNAREs in leaves at 12 hours and 22 hours post-inoculation with S. sclerotiorum; Table S11: Expression profiles of Qa-SNAREs in resistant (Zhongshuang9) and susceptible (84039) varieties of B. napus under treatments with ABA, MeJA, SA, and OA; Table S12: Putative protein–protein interaction networks among BnaSYP121s, BnaSYP122s, BnaSNAPs, BnaVAMP721s, and BnaVAMP722/3s; Table S13: List of AtSNAREs interacting proteins predicted by STRING-DB; Table S14: Orthologs of AtSNAREs interacting proteins in Brassica napus; Table S15: Primers used in this study.
Author Contributions: Conceptualization, J.X., J.B., and A.W.; data curation, J.X. Y.S. (Yanan Shan), M.Z., and Y.S. (Yanan Shen); formal analysis, J.X.; investigation, J.X., X.Z., and Y.S. (Yanan Shan); methodology, J.X.; project administration, A.W.; resources, J.X., X.Z., and A.W.; software, J.X.; supervision, G.L., Z.W., and A.W.; validation, J.X. and A.W.; visualization, J.X.; writing—original draft, J.X.; writing—review and editing, J.X., Y.S.A., and A.W. All authors have read and agreed to the published version of the manuscript.
: the National Natural Science Foundation of China (Grant Nos. 31972353).
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: All data generated or analyzed during this study are included in this article and its Supplementary Information Files. The RNA-Seq data libraries were generated in this research. The sequences of B. napus and expression profiles of BnaSNAREs downloaded from the B. napus genome resources are accessible via the following link https://.genoscope.cns.fr/brassicanapus/, accessed on 26 October 2020.
: This work was supported by the National Natural Science Foundation of China (Grant Nos. 31972353). We thank Shengyi Liu (Oil Crops Research Institute, Chinese Academy of Agricultural Sciences) for providing the seeds of Zhongshuang9 and 84039 and Martin B. Dickman for providing S. sclerotiorum 1980.
Conflicts of Interest: The authors declare that they have no competing interests. All authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. https://doi.org/10.1111/tpj.13669.
- Bayer, P.E.; Hurgobin, B.; Golicz, A.A.; Chan, C.-K.K.; Yuan, Y.; Lee, H.; Renton, M.; Meng, J.; Li, R.; Long, Y.; et al. Assembly and comparison of two closely related Brassica napus genomes. Plant Biotechnol. J. 2017, 15, 1602–1610. https://doi.org/10.1111/pbi.12742.
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005, 8, 135–141. https://doi.org/10.1016/j.pbi.2005.01.001.
- Cui, L.Y.; Wall, P.K.; Leebens-Mack, J.H.; Lindsay, B.G.; Soltis, D.E.; Doyle, J.J.; Soltis, P.S.; Carlson, J.E.; Arumuganathan, K.; Barakat, A.; et al. Widespread genome duplications throughout the history of flowering plants. Genome Res. 2006, 16, 738–749. https://doi.org/10.1101/gr.4825606.
- Grassi, A. de; Lanave, C.; Saccone, C. Genome duplication and gene-family evolution: The case of three OXPHOS gene fami-lies. Gene 2008, 421, 1–6. https://doi.org/10.1016/j.gene.2008.05.011.
- Zhao, W.; Dong, S.; Ye, W.; Hua, C.; Meijer, H.J.G.; Dou, X.; Govers, F.; Wang, Y. Genome-wide identification of Phytophthora sojae SNARE genes and functional characterization of the conserved SNARE PsYKT6. Fungal Genet. Biol. 2011, 48, 241–251. https://doi.org/10.1016/j.fgb.2010.11.006.
- Cucu, B.; Degreif, D.; Bertl, A.; Thiel, G. Vesicle fusion and fission in plants and yeast. Cell Calcium 2017, 67, 40–45. https://doi.org/10.1016/j.ceca.2017.08.007.
- Bonifacino, J.S.; Glick, B.S. The mechanisms of vesicle budding and fusion. Cell 2004, 116, 153–166. https://doi.org/10.1016/s0092-8674(03)01079-1.
- Söllner, T.; Bennett, M.K.; Whiteheart, S.W.; Scheller, R.H.; Rothman, J.E. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 1993, 75, 409–418. https://doi.org/10.1016/0092-8674(93)90376-2.
- Fasshauer, D.; Sutton, R.B.; Brunger, A.T.; Jahn, R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. USA 1998, 95, 15781–15786. https://doi.org/10.1073/pnas.95.26.15781.
- Sanderfoot, A. Increases in the number of SNARE genes parallels the rise of multicellularity among the green plants. Plant Physiol. 2007, 144, 6–17. https://doi.org/10.1104/pp.106.092973.
- Salinas-Cornejo, J.; Madrid-Espinoza, J.; Ruiz-Lara, S. Identification and transcriptional analysis of SNARE vesicle fusion regulators in tomato (Solanum lycopersicum) during plant development and a comparative analysis of the response to salt stress with wild relatives. J. Plant Physiol. 2019, 242, 153018. https://doi.org/10.1016/j.jplph.2019.153018.
- Sansebastiano, G.D.; Piro, G. The SNARE proteins (in plants) beyond the Nobel Prize. J. Plant Biochem. Physiol. 2014, 2, e122. https://doi.org/10.4172/2329-9029.1000e122.
- Wang, G.; Long, D.; Yu, F.; Zhang, H.; Chen, C.; Wang, Y.; Ji, W. Genome-wide identification, evolution, and expression of the SNARE gene family in wheat resistance to powdery mildew. PeerJ 2021, 9, e10788. https://doi.org/10.7717/peerj.10788.
- Rosenbaum, E.E.; Vasiljevic, E.; Cleland, S.C.; Flores, C.; Colley, N.J. The Gos28 SNARE protein mediates intra-Golgi transport of rhodopsin and is required for photoreceptor survival. J. Biol. Chem. 2014, 289, 32392–32409. https://doi.org/10.1074/jbc.M114.585166.
- Kienle, N.; Kloepper, T.H.; Fasshauer, D. Differences in the SNARE evolution of fungi and metazoa. Biochem. Soc. Trans. 2009, 37, 787–791. https://doi.org/10.1042/BST0370787.
- Burri, L.; Lithgow, T. A complete set of SNAREs in yeast. Traffic 2004, 5, 45–52. https://doi.org/10.1046/j.1600-0854.2003.00151.x.
- Furukawa, N.; Mima, J. Multiple and distinct strategies of yeast SNAREs to confer the specificity of membrane fusion. Sci. Rep. 2014, 4, 4277. https://doi.org/10.1038/srep04277.
- Kim, S. J.; Brandizzi, F. News and views into the SNARE complexity in Arabidopsis. Front. Plant Sci. 2012, 3, 28. https://doi.org/10.3389/fpls.2012.00028.
- Ben, Z.; Karnik, R.; YiZhou, W.; Wallmeroth, N.; Blatt, M.R.; Grefen, C. The Arabidopsis R-SNARE VAMP721 interacts with KAT1 and KC1 K+ channels to moderate K+ current at the plasma membrane. Plant Cell 2015, 27, 1697–1717. https://doi.org/10.1105/tpc.15.00305.
- Uemura, T.; Kim, H.; Saito, C.; Ebine, K.; Ueda, T.; Schulze-Lefert, P.; Nakano, A. Qa-SNAREs localized to the trans-Golgi network regulate multiple transport pathways and extracellular disease resistance in plants. Proc. Natl. Acad. Sci. USA 2012, 109, 1784–1789. https://doi.org/10.1073/pnas.1115146109.
- Chung, K.P.; Zeng, Y.; Li, Y.; Ji, C.; Xia, Y.; Jiang, L. Signal motif-dependent ER export of the Qc-SNARE BET12 interacts with MEMB12 and affects PR1 trafficking in Arabidopsis. J. Cell Sci. 2018, 131, jcs202838. https://doi.org/10.1242/jcs.202838.
- Lauber, M.H.; Waizenegger, I.; Steinmann, T.; Schwarz, H.; Mayer, U.; Hwang, I.; Lukowitz, W.; Jürgens, G. The Arabidopsis KNOLLE protein is a cytokinesis-specific syntaxin. J. Cell Biol. 1997, 139, 1485–1493. https://doi.org/10.1083/jcb.139.6.1485.
- Slane, D.; Reichardt, I.; El Kasmi, F.; Bayer, M.; Jürgens, G. Evolutionarily diverse SYP1 Qa-SNAREs jointly sustain pollen tube growth in Arabidopsis. Plant J. 2017, 92, 375–385. https://doi.org/10.1111/tpj.13659.
- Lian, Q.; Meng, Y.; Zhao, X.; Xu, Y.; Wang, Y.; Day, B.; Ma, Q. ShNPSN11, a vesicle-transport-related gene, confers disease resistance in tomato to Oidium neolycopersici. Biochem. J. 2020, 477, 3851–3866. https://doi.org/10.1042/BCJ20190776.
- Nühse, T.S.; Boller, T.; Peck, S.C. A plasma membrane syntaxin is phosphorylated in response to the bacterial elicitor flagel-lin. J. Biol. Chem. 2003, 278, 45248–45254. https://doi.org/10.1074/jbc.M307443200.
- Assaad, F.F.; Qiu, J.-L.; Youngs, H.; Ehrhardt, D.; Zimmerli, L.; Kalde, M.; Wanner, G.; Peck, S.C.; Edwards, H.; Ramonell, K.; et al. The PEN1 syntaxin defines a novel cellular compartment upon fungal attack and is required for the timely assembly of papillae. Mol. Biol. Cell 2004, 15, 5118–5129. https://doi.org/10.1091/mbc.e04-02-0140.
- Zhang, Z.; Feechan, A.; Pedersen, C.; Newman, M.-A.; Qiu, J.-L.; Olesen, K.L.; Thordal-Christensen, H. A SNARE-protein has opposing functions in penetration resistance and defence signalling pathways. Plant J. 2007, 49, 302–312. https://doi.org/10.1111/j.1365-313X.2006.02961.x.
- Catalano, C.M.; Czymmek, K.J.; Gann, J.G.; Sherrier, D.J. Medicago truncatula syntaxin SYP132 defines the symbiosome membrane and infection droplet membrane in root nodules. Planta 2007, 225, 541–550. https://doi.org/10.1007/s00425-006-0369-y.
- Kalde, M.; Nühse, T.S.; Findlay, K.; Peck, S.C. The syntaxin SYP132 contributes to plant resistance against bacteria and secre-tion of pathogenesis-related protein 1. Proc. Natl. Acad. Sci. USA 2007, 104, 11850–11855. https://doi.org/10.1073/pnas.0701083104.
- Zhu, J.; Gong, Z.; Zhang, C.; Song, C.-P.; Damsz, B.; Inan, G.; Koiwa, H.; Zhu, J.-K.; Hasegawa, P.M.; Bressan, R.A. OSM1/SYP61: A syntaxin protein in Arabidopsis controls abscisic acid-mediated and non-abscisic acid-mediated responses to abiotic stress. Plant Cell 2002, 14, 3009–3028. https://doi.org/10.1105/tpc.006981.
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. https://doi.org/10.1126/science.1253435.
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. https://doi.org/10.1111/tpj.13669.
- Bayer, P.E.; Hurgobin, B.; Golicz, A.A.; Chan, C.-K.K.; Yuan, Y.; Lee, H.; Renton, M.; Meng, J.; Li, R.; Long, Y.; et al. Assembly and comparison of two closely related Brassica napus genomes. Plant Biotechnol. J. 2017, 15, 1602–1610. https://doi.org/10.1111/pbi.12742.
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005, 8, 135–141. https://doi.org/10.1016/j.pbi.2005.01.001.
- Cui, L.Y.; Wall, P.K.; Leebens-Mack, J.H.; Lindsay, B.G.; Soltis, D.E.; Doyle, J.J.; Soltis, P.S.; Carlson, J.E.; Arumuganathan, K.; Barakat, A.; et al. Widespread genome duplications throughout the history of flowering plants. Genome Res. 2006, 16, 738–749. https://doi.org/10.1101/gr.4825606.
- Grassi, A. de; Lanave, C.; Saccone, C. Genome duplication and gene-family evolution: The case of three OXPHOS gene fami-lies. Gene 2008, 421, 1–6. https://doi.org/10.1016/j.gene.2008.05.011.