 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Xiawei Dang | -- | 2150 | 2022-04-27 23:49:31 | | | |
| 2 | Dean Liu | Meta information modification | 2150 | 2022-04-28 04:18:29 | | | | |
| 3 | Dean Liu | Meta information modification | 2150 | 2022-04-29 07:42:01 | | |
Video Upload Options
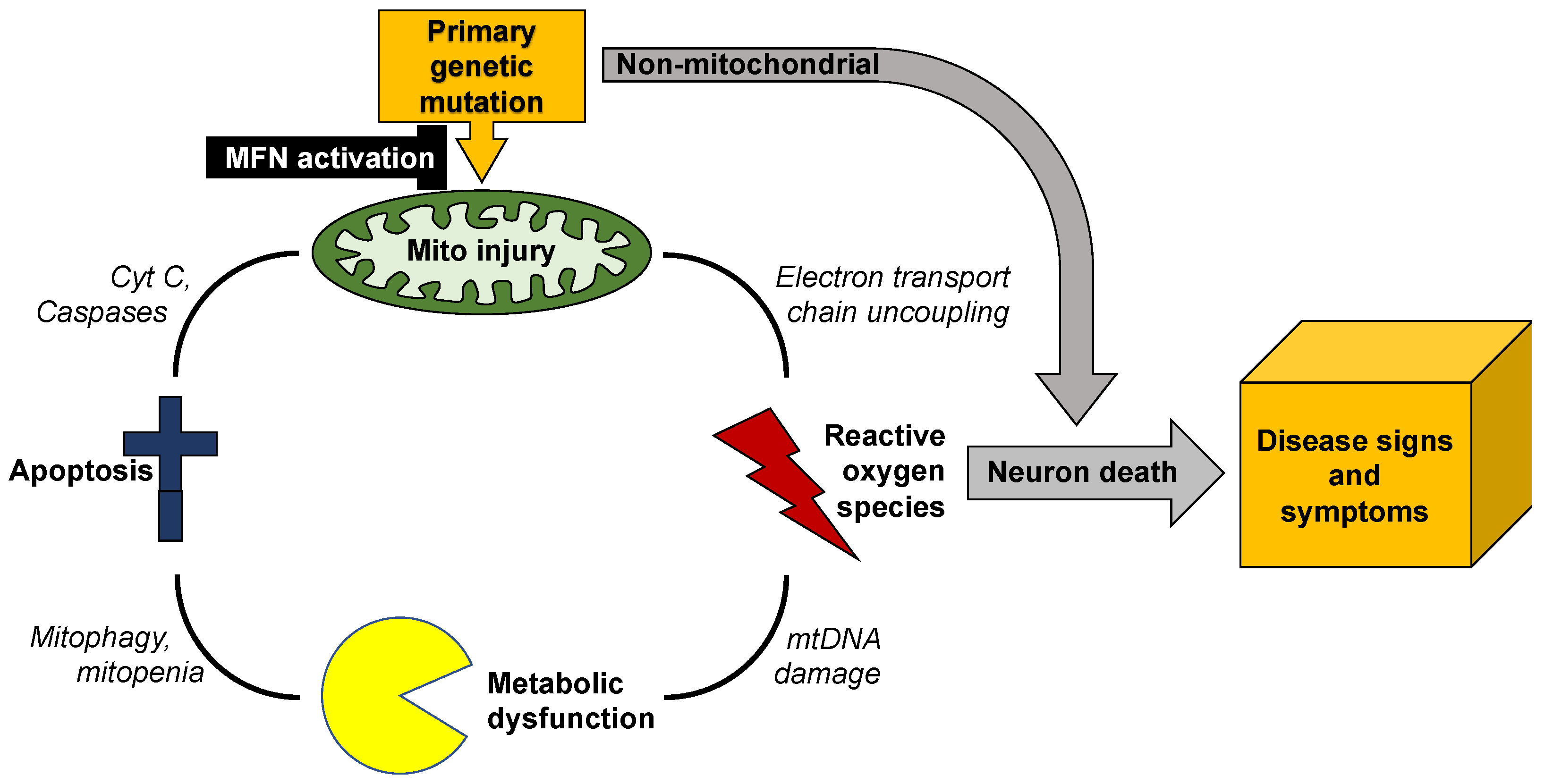
Mitochondrial dynamics encompass mitochondrial fusion, fission, and movement. Mitochondrial fission and fusion are seemingly ubiquitous, whereas mitochondrial movement is especially important for organelle transport through neuronal axons. In addition to review mitochondrial dynamics processes in Charcot–Marie–Tooth disease type 2A, amyotrophic lateral sclerosis, Friedrich’s ataxia, dominant optic atrophy, Alzheimer’s, Huntington’s and Parkinson’s diseases, researchers suggest a possible screening procedure using patient-derived cells to evaluate different therapeutics for a given disease, or to assess potential efficacy of a particular approach in multiple diseases.
1. Introduction
2. Mitochondrial Dysdynamism in Genetic Neurological Diseases
3. Evaluating Mitochondrial Dynamics
4. Summary and Conclusions
Mitochondrial dynamism is ubiquitous, but the relative rates and importance of mitochondrial fusion, fission, and motility differ according to cell morphology and metabolic requirements. Thus, neurons with high metabolic requirements and long axons have unique dynamic profiles and are especially susceptible to dynamic dysfunction. Mitochondrial phenotypes are difficult to evaluate in human neurodegenerative diseases because human neurons are not readily available.
Previous and accompanying data demonstrate that cultured human dermal fibroblasts can be induced to exhibit neurodegenerative disease-related mitochondrial fusion-fission phenotypes by forcing mitochondrial metabolism through the simple maneuver of substituting galactose for glucose in the culture medium. Moreover, the same fibroblasts can be directly reprogrammed into neurons that retain hallmark mitochondrial abnormalities, including dysmotility, which is best measured in neuronal axons. Researchers suggest that integration of these platforms with genetic mouse models, as depicted in Figure 2, might be an effective means of evaluating candidate therapeutics in the multitude of genetic neurodegenerative conditions exhibiting characteristic mitochondrial abnormalities, or of different patients for a personalized medicine approach.

Figure 2. Possible screening procedure using patient-derived cells to evaluate different therapeutics for a given disease, or to assess potential efficacy of a particular approach in multiple diseases.
References
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176.
- Dorn, G.W., II. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17.
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 2016, 21, 1327–1335.
- Song, M.; Dorn, G.W., II. Mitoconfusion: Noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 2015, 21, 195–205.
- Dorn, G.W., II. Mitochondrial dynamism and heart disease: Changing shape and shaping change. EMBO Mol. Med. 2015, 7, 865–877.
- MacAskill, A.F.; Kittler, J.T. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010, 20, 102–112.
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93.
- Pareyson, D.; Piscosquito, G.; Moroni, I.; Salsano, E.; Zeviani, M. Peripheral neuropathy in mitochondrial disorders. Lancet Neurol. 2013, 12, 1011–1024.
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of central nervous system to body metabolism in vertebrates: Its constancy and functional basis. Am. J. Physiol. 1981, 241, R203–R212.
- Ashrafi, G.; Ryan, T.A. Glucose metabolism in nerve terminals. Curr. Opin. Neurobiol. 2017, 45, 156–161.
- Baloh, R.H.; Schmidt, R.E.; Pestronk, A.; Milbrandt, J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J. Neurosci. 2007, 27, 422–430.
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728.
- De Vos, K.J.; Grierson, A.J.; Ackerley, S.; Miller, C.C. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 2008, 31, 151–173.
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518.
- Guo, X.; Disatnik, M.H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Investig. 2013, 123, 5371–5388.
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2018, 9, 6128–6143.
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, 1–17.
- Marchesi, C.; Ciano, C.; Salsano, E.; Nanetti, L.; Milani, M.; Gellera, C.; Taroni, F.; Fabrizi, G.M.; Uncini, A.; Pareyson, D. Co-occurrence of amyotrophic lateral sclerosis and Charcot-Marie-Tooth disease type 2A in a patient with a novel mutation in the mitofusin-2 gene. Neuromuscul. Disord. 2011, 21, 129–131.
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719.
- Khalil, B.; Cabirol-Pol, M.J.; Miguel, L.; Whitworth, A.J.; Lecourtois, M.; Lievens, J.C. Enhancing Mitofusin/Marf ameliorates neuromuscular dysfunction in Drosophila models of TDP-43 proteinopathies. Neurobiol. Aging 2017, 54, 71–83.
- Baek, M.; Choe, Y.J.; Bannwarth, S.; Kim, J.; Maitra, S.; Dorn, G.W., II; Taylor, J.P.; Paquis-Flucklinger, V.; Kim, N.C. TDP-43 and PINK1 mediate CHCHD10(S59L) mutation-induced defects in Drosophila and in vitro. Nat. Commun. 2021, 12, 1924.
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065.
- Gilkerson, R.W.; Schon, E.A.; Hernandez, E.; Davidson, M.M. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J. Cell Biol. 2008, 181, 1117–1128.
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 1–17.
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477.
- Dorn, G.W., II. Mitofusin 2 Dysfunction and Disease in Mice and Men. Front. Physiol. 2020, 11, 782.
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044.
- Sah, E.; Krishnamurthy, S.; Ahmidouch, M.Y.; Gillispie, G.J.; Milligan, C.; Orr, M.E. The Cellular Senescence Stress Response in Post-Mitotic Brain Cells: Cell Survival at the Expense of Tissue Degeneration. Life 2021, 11, 229.
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451.
- Tan, C.A.; Rabideau, M.; Blevins, A.; Westbrook, M.J.; Ekstein, T.; Nykamp, K.; Deucher, A.; Harper, A.; Demmer, L. Autosomal recessive MFN2-related Charcot-Marie-Tooth disease with diaphragmatic weakness: Case report and literature review. Am. J. Med. Genet. A 2016, 170, 1580–1584.
- Bombelli, F.; Stojkovic, T.; Dubourg, O.; Echaniz-Laguna, A.; Tardieu, S.; Larcher, K.; Amati-Bonneau, P.; Latour, P.; Vignal, O.; Cazeneuve, C.; et al. Charcot-Marie-Tooth disease type 2A: From typical to rare phenotypic and genotypic features. JAMA Neurol. 2014, 71, 1036–1042.
- Pipis, M.; Feely, S.M.E.; Polke, J.M.; Skorupinska, M.; Perez, L.; Shy, R.R.; Laura, M.; Morrow, J.M.; Moroni, I.; Pisciotta, C.; et al. Natural history of Charcot-Marie-Tooth disease type 2A: A large international multicentre study. Brain 2020, 143, 3589–3602.
- Franco, A.; Dang, X.; Walton, E.K.; Ho, J.N.; Zablocka, B.; Ly, C.; Miller, T.M.; Baloh, R.H.; Shy, M.E.; Yoo, A.S.; et al. Burst mitofusin activation reverses neuromuscular dysfunction in murine CMT2A. eLife 2020, 9, e61119.
- Zhou, Y.; Carmona, S.; Muhammad, A.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W., II; et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Investig. 2021, 13, e147307.
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R. H, Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240.
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341.
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250.
- Wang, H.; Lim, P.J.; Karbowski, M.; Monteiro, M.J. Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum. Mol. Genet. 2009, 18, 737–752.
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23.
- Ligon, L.A.; LaMonte, B.H.; Wallace, K.E.; Weber, N.; Kalb, R.G.; Holzbaur, E.L. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport 2005, 16, 533–536.
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16.
- Bates, G.P. History of genetic disease: The molecular genetics of Huntington disease—A history. Nat. Rev. Genet. 2005, 6, 766–773.
- Polyzos, A.A.; McMurray, C.T. The chicken or the egg: Mitochondrial dysfunction as a cause or consequence of toxicity in Huntington’s disease. Mech. Ageing Dev. 2017, 161, 181–197.
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215.
- Yu-Wai-Man, P.; Chinnery, P.F. Dominant optic atrophy: Novel OPA1 mutations and revised prevalence estimates. Ophthalmology 2013, 120, 1712–1712.e1.
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46.
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240.
- Cretin, E.; Lopes, P.; Vimont, E.; Tatsuta, T.; Langer, T.; Gazi, A.; Sachse, M.; Yu-Wai-Man, P.; Reynier, P.; Wai, T. High-throughput screening identifies suppressors of mitochondrial fragmentation in OPA1 fibroblasts. EMBO Mol. Med. 2021, 13, e13579.
- Zanna, C.; Ghelli, A.; Porcelli, A.M.; Karbowski, M.; Youle, R.J.; Schimpf, S.; Wissinger, B.; Pinti, M.; Cossarizza, A.; Vidoni, S.; et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2008, 131, 352–367.
- Gerber, S.; Charif, M.; Chevrollier, A.; Chaumette, T.; Angebault, C.; Kane, M.S.; Paris, A.; Alban, J.; Quiles, M.; Delettre, C.; et al. Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain 2017, 140, 2586–2596.
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30.
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323.
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10.
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273.
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839.
- Pandey, A.; Gordon, D.M.; Pain, J.; Stemmler, T.L.; Dancis, A.; Pain, D. Frataxin directly stimulates mitochondrial cysteine desulfurase by exposing substrate-binding sites, and a mutant Fe-S cluster scaffold protein with frataxin-bypassing ability acts similarly. J. Biol. Chem. 2013, 288, 36773–36786.
- Sahdeo, S.; Scott, B.D.; McMackin, M.Z.; Jasoliya, M.; Brown, B.; Wulff, H.; Perlman, S.L.; Pook, M.A.; Cortopassi, G.A. Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich’s ataxia. Hum. Mol. Genet. 2014, 23, 6848–6862.
- Stepanova, A.; Magrané, J. Mitochondrial dysfunction in neurons in Friedreich’s ataxia. Mol. Cell. Neurosci. 2020, 102, 103419.
- Kanazawa, M.; Yano, M.; Namchai, C.; Yamamoto, S.; Ohtake, A.; Takayanagi, M.; Mori, M.; Niimi, H. Visualization of mitochondria with green fluorescent protein in cultured fibroblasts from patients with mitochondrial diseases. Biochem. Biophys. Res. Commun. 1997, 239, 580–584.
- Pham, N.A.; Richardson, T.; Cameron, J.; Chue, B.; Robinson, B.H. Altered mitochondrial structure and motion dynamics in living cells with energy metabolism defects revealed by real time microscope imaging. Microsc. Microanal. 2004, 10, 247–260.
- Kuznetsov, A.V.; Hermann, M.; Saks, V.; Hengster, P.; Margreiter, R. The cell-type specificity of mitochondrial dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 1928–1939.
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79.
- Pham, A.H.; McCaffery, J.M.; Chan, D.C. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis 2012, 50, 833–843.

