1. Introduction
Mitochondrial dynamics include mitochondrial fission, fusion, and motility [
4,
5]. At the observational level, these are distinct processes whose activity is determined by cell type and pathophysiological context. Thus, mitochondria in quiescent fibroblasts are part of highly interconnected networks wherein mitochondrial fusion and fission mediate continuous structural remodeling of the collective; directed subcellular transport of individual mitochondria is infrequent (~2%–4%). However, prior to fibroblast mitosis and cytokinesis, the mitochondrial network undergoes fission-mediated fragmentation, enabling around half of the resulting individual organelles to be directed into each of the daughter cell precursors [
6]. By contrast, mitochondria of cardiac and skeletal myocytes are seemingly static groups of individual organelles stationed between myofilaments; fusion and fission are infrequent [
7,
8]. In neurons, mitochondrial dynamism proceeds according to subcellular location: mitochondria in neuronal soma exist as stationary perinuclear clusters, mitochondria within neuronal processes are either anchored in small clusters (~70%) or are actively undergoing antegrade or retrograde transport (~30%), and mitochondria within synaptic neuromuscular junctions tend to be stationary individual organelles [
9,
10]. The diversity of mitochondrial structure and differential application of mitochondrial dynamics according to cell type and subcellular location may explain why mitochondrial dynamic dysfunction most severely impacts the neurological system [
11]
2. Mitochondrial Dysdynamism in Genetic Neurological Diseases
Conceptually, it is easy to appreciate how mitochondrial dynamic dysfunction can manifest as neurodegenerative disease. Electrically excitable neurons have high metabolic requirements to maintain electrochemical membrane potentials and drive synaptic release and reuptake of neurotransmitters. Indeed, the neurological system has one of the highest metabolic rates of all organ systems; human brains, which comprise only ~2% of total body weight, butconsume ~20% of total oxygen (a marker for ATP production by mitochondrial respiration) [
35,
36]. Therefore, any genetic or environmental event that compromises mitochondrial fitness has the potential to adversely impact the neurological system.
In addition to high metabolic needs, neurons have a unique structural feature making them exceptionally vulnerable to mitochondrial dysfunction: neurons are long. Sciatic nerve motor and sensory neurons originating in the lumbar spine of a six-foot-tall person may extend ~three feet before they terminate in the feet. Mitochondrial-derived ATP is required throughout the neuron, and because ATP undergoes rapid spontaneous degradation by hydrolysis, it must be generated locally by resident mitochondria according to physiological need. The length of neuronal processes presents a formidable physical obstacle to effective mitochondrial delivery. For this reason, neurons (especially the long peripheral nerves innervating upper and lower extremities) are susceptible to damage from factors that diminish mitochondrial transport [
37,
38,
39].
Mitochondrial dysmotility is a common finding in human genetic neurodegenerative diseases, especially peripheral neuropathies (
vide infra). The most common mitochondrial abnormality in these syndromes is mitochondrial morphological shortening, or fragmentation. In most conditions the underlying abnormality in mitochondrial dynamics is increased DRP1-mediated mitochondrial fission [
40,
41,
42,
43]. However, an impairment of MFN-mediated mitochondrial fusion is increasingly recognized as an accompanying or alternative mechanism for observed mitochondrial fragmentation [
44,
45,
46,
47]. This is an important distinction because increased mitochondrial fission does not necessarily provoke mitochondrial dysfunction, whereas suppressing mitochondrial fusion interrupts complementation-mediated mitochondrial repair and causes respiratory/metabolic dysfunction (
vide supra) [
21,
22]. Moreover, mitochondrial respiratory dysfunction is damaging both because it impedes ATP production and because uncoupling of oxidative phosphorylation from ATP synthesis produces neuro- and mito-toxic reactive oxygen species (ROS). Thus, mitochondrial dysdynamism can initiate a vicious cycle of mitochondrial degeneration in which a primary impairment in the ability of individual mitochondria to undergo fusion-mediated repair evokes accelerated organelle senescence manifested in part by increased production of toxic ROS, which further injures the index organelle and can damage other members of the mitochondrial collective, which have similar dynamic impairment. Mitochondrial damage provokes more mitochondrial damage (
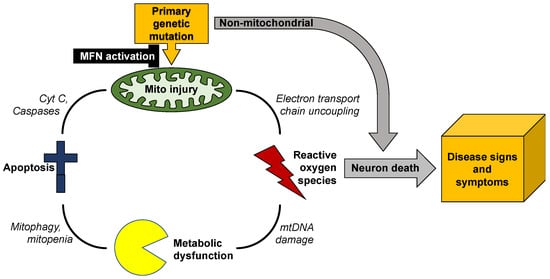
Figure 3).
Figure 3. Vicious cycle of mitochondrial degeneration in neurodegenerative diseases.
Unless this feed-forward cycle is interrupted, mitochondrial degeneration can spread throughout the neuron, depriving it of ATP required for synaptic transmission and neuronal repair, and exposing it to ROS that will degrade nuclear DNA and damage essential proteins [
48]. Because mitochondrial dysmotility and degeneration tend to be most severe distally, the ultimate consequence is neuronal die-back [
49,
50] or, if mitochondrial damage is more uniform, apoptotic neuron death [
51,
52]. This paradigm can explain why such genetically and etiologically diverse neurodegenerative diseases as amyotrophic lateral sclerosis, Huntington’s disease, and Charcot–Marie–Tooth disease share common phenotypes of mitochondrial degeneration and neuron loss.
Charcot–Marie–Tooth disease type 2A (CMT2A) is the only neurodegenerative syndrome unambiguously caused by mitochondrial dynamic dysfunction. This slowly progressive pediatric peripheral neuropathy is linked to ~100 different loss-of-function mutations of the
MFN2 gene that encodes one of the two outer mitochondrial membrane mitofusin proteins [
50,
53]. Clinically, autosomal dominant CMT2A typically manifests in toddlers as difficulty walking and delayed progress in ambulation; rare autosomal recessive CMT2A has a similar presentation [
54]. The disease progresses during childhood and adolescence, culminating in disabling neurogenic muscular atrophy of the distal upper and lower extremities most often requiring wheelchairs for mobility and hand and foot splints to prevent secondary skeletal abnormalities [
55,
56]. The underlying neuropathology is progressive neuronal “die-back”, described as retrograde degeneration from the distal synapses of long peripheral motor and sensory nerves. In most instances die-back stops at approximately the knee and elbow, producing characteristic distal neurogenic muscle atrophy that spares proximal extremities. Axonal mitochondria in (directly reprogrammed) human CMT2A neurons or neurons from mice expressing human CMT2A MFN2 mutants exhibit fragmentation, depolarization (dissipated ΔΨm), and severe hypomotility [
57]. Mitochondria in CMT2A neuronal soma are abnormally aggregated, forming characteristic “clumps” [
58], and mitochondria can be lost from CMT2A neuronal neuromuscular junctions [
57,
58]. Mitochondrial fragmentation in CMT2A is the direct effect of dominant suppression of mitochondrial fusion by MFN2 mutants, while loss of inner membrane ΔΨm is likely a secondary effect of impaired fusion-mediated mitochondrial repair (
vide supra). The exact mechanism for mitochondrial dysmotility in CMT2A is unclear, although there is evidence that MFN2 normally binds to and may therefore modulate mitochondrial Miro that couples mitochondria to the Milton/Trak transport apparatus [
59]. Pharmacological activation of endogenous normal MFN1 and MFN2 (which appear functionally overlapping for mitochondrial fusion and motility), can improve mitochondrial abnormalities produced by CMT2A MFN2 mutants in neurons [
25] and reverse neuromuscular degeneration in CMT2A mice [
57].
Abnormalities in mitochondrial morphology and mitofusin expression/function are equally prominent in amyotrophic lateral sclerosis (ALS) [
38,
60] and Huntington’s disease [
41,
61], although the primary genetic causes of these diseases are not mutations in mitochondrial genes. ALS is an etiologically diverse upper and lower motor neuron neuropathy. Familial ALS with an identifiable genetic cause occurs in 1 out of every 10 cases [
62], and mitochondrial fragmentation and impaired mitochondrial transport are typical [
38,
60,
63,
64]. By contrast, Huntington’s disease (HD) is a central nervous system cognitive and motor disorder caused by an expanded number of CAG repeats in the
huntingtin (
HTT) gene [
65]. Mitochondrial fragmentation and depolarization are hallmarks of this condition [
41], although it is not clear whether the mitochondrial abnormalities are principal contributing factors that provoke neuronal death, or reflect collateral damage that is a consequence of other primary abnormalities [
66].
Mutations of the inner mitochondrial membrane fusion protein, optic atrophy 1 (OPA1), cause dominant optic atrophy (DOA) in approximately half of cases, primarily affecting retinal ganglion cells [
67,
68,
69]. An excess of mitochondrial fission relative to fusion in retinal ganglion cell axons from OPA1 mutant mice and patient fibroblasts is evidenced by prototypical mitochondrial fragmentation [
70,
71,
72]. Mutations of dynamin-like protein 1, which is a central effector of mitochondrial fission, are also linked to DOA. In contrast to OPA1 mutations that interrupt mitochondrial fusion,
DNM1L mutations produce a relative excessive of mitochondrial fusion verses fission, with elongated/tubular mitochondria in patient-derived fibroblasts [
20]. Thus, the balance between mitochondrial fusion and fission, rather than the absolute activity of either process, appears critical to neuronal health.
Mitochondria are not abnormal in all genetic neurodegenerative diseases, and mitochondrial dysdynamism is not necessarily a component of all neurodegenerative conditions that exhibit mitochondrial abnormalities. For example, metabolic abnormalities are described in Alzheimer’s dementia [
73], a disease of uncertain and complex etiology linked to formation of extracellular deposits of β-amyloid peptide and neurofibrillary tangles of microtubular tau protein [
74]. Reported mitochondrial defects, which include organelle fragmentation, are varied and inconsistent [
73,
75]. The sheer scope of different mitochondrial abnormalities described in Alzheimer’s disease confounds conclusions about whether they contribute in a meaningful way to, or are largely a consequence of, the primary disease. By contrast, heritable Parkinson’s disease is clearly a disease of mitochondria, since the most common genetic forms of Parkinsonism are linked to mutations of genes encoding mitochondrial PINK1 kinase (
PARK6) that initiates mitophagy, as well as the principal mitophagy effector protein, Parkin (
PARK2) [
76]. Again, mitochondrial fragmentation is reported in some, but not all, descriptions of Parkinson’s disease [
77], likely because of heterogeneity and multifactorial etiology. Finally, Friedreich’s ataxia/spinocerebellar degeneration is unambiguously a neurodegenerative disease with a mitochondrial etiology; the autosomal recessive genetic defect is in the
FXN gene that encodes the mitochondrial iron-binding protein, frataxin. Mitochondrial iron overload in Friedreich’s ataxia compromises ATP formation by the electron transport chain and provokes excessive ROS elaboration [
78,
79]. It is not clear if mitochondrial dynamic dysfunction contributes to, or even exists, in this disease [
80]. Variable mitochondrial phenotypes in neurodegenerative diseases and uncertainty regarding the pathophysiological impact of abnormal mitochondrial dynamics emphasize the need for analytical platforms directly relevant to individual human patients.
3. Evaluating Mitochondrial Dynamics
The most readily obtained and obvious evidence for mitochondrial dysdynamism is abnormal mitochondrial morphology in static images of live or fixed cells, typically imaged with a mitochondrial specific fluorophore. The most common reported mitochondrial structural abnormality is “fragmentation” or structural shortening of the organelles, conventionally and conveniently measured as reduced aspect ratio (length/width) [
81,
82]. Although the “normal” mitochondrial aspect ratio depends both upon cell type and physiological status [
83], measuring it is conceptually and technically straightforward. Thus, a decrease in mitochondrial aspect ratio observed in high-resolution images (reduced length/width or shortening) typically reflects a relative increase in mitochondrial fission to fusion (“fragmentation”), whereas an increase in aspect ratio (“elongation”) reflects an increase in fusion relative to fission. However, altered aspect ratio does not reveal the underlying cause of dysdynamism, i.e., whether the dynamic imbalance is the consequence of altered fission, or fusion, or both. Moreover, fragmentation of mitochondria in cells having interconnected mitochondrial networks, such as fibroblasts, can be a normal component of mitosis and apoptosis [
6]. Finally, short, spherical, or “fragmented” mitochondria are the norm in striated muscle cells and neuronal axons [
7,
8,
83].
The nature of mitochondrial fusion/fission disequilibrium is best revealed by live cell imaging, using either a single mitochondrial fluorophore (e.g., Mitotracker Red, Green, or Orange), fused cells expressing different mitochondrial-targeted fluorescent proteins (e.g., Mito-GFP and Mito-RFP), or a photo-switchable mitochondrial fluorophore (e.g., MitoDendra). When a single fluorophore is used, video-confocal microscopy can document mitochondrial fusion and fission events [
82]. When different groups of cells expressing mitochondrial targeted GFP or RFP are fused with polyethylene glycol, it becomes possible to quantify mitochondria content mixing from fusion as a function of time [
84]. This same approach is easier using photo-switchable mito-Dendra [
85], enabling a subset of mitochondria within cells of interest to be photo-switched from green to red fluorescing, and the fate of individual mitochondria (i.e., whether they are fusing, undergoing fission, or being transported within the cell) to be assessed over time. However, the photo-switching approach requires special equipment and is limited by sample throughput.
- Summary and Conclusions
Mitochondrial dynamism is ubiquitous, but the relative rates and importance of mitochondrial fusion, fission, and motility differ according to cell morphology and metabolic requirements. Thus, neurons with high metabolic requirements and long axons have unique dynamic profiles and are especially susceptible to dynamic dysfunction. Mitochondrial phenotypes are difficult to evaluate in human neurodegenerative diseases because human neurons are not readily available.
Previous and accompanying data demonstrate that cultured human dermal fibroblasts can be induced to exhibit neurodegenerative disease-related mitochondrial fusion-fission phenotypes by forcing mitochondrial metabolism through the simple maneuver of substituting galactose for glucose in the culture medium. Moreover, the same fibroblasts can be directly reprogrammed into neurons that retain hallmark mitochondrial abnormalities, including dysmotility, which is best measured in neuronal axons. We suggest that integration of these platforms with genetic mouse models, as depicted in Figure 4, might be an effective means of evaluating candidate therapeutics in the multitude of genetic neurodegenerative conditions exhibiting characteristic mitochondrial abnormalities, or of different patients for a personalized medicine approach.

Figure 4. Possible screening procedure using patient-derived cells to evaluate different therapeutics for a given disease, or to assess potential efficacy of a particular approach in multiple diseases.
This entry is adapted from the peer-reviewed paper 10.3390/cells11061049