Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | MADHAVAN NAMPOOTHIRI G | -- | 3381 | 2022-04-26 11:19:01 | | | |
| 2 | Conner Chen | Meta information modification | 3381 | 2022-04-27 04:01:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nampoothiri G, M.; , .; Gurram, P.C.; Mudgal, J. Astrocytic Glutamatergic Transmission in Neurodegenerative Disorders. Encyclopedia. Available online: https://encyclopedia.pub/entry/22289 (accessed on 24 June 2026).
Nampoothiri G M, , Gurram PC, Mudgal J. Astrocytic Glutamatergic Transmission in Neurodegenerative Disorders. Encyclopedia. Available at: https://encyclopedia.pub/entry/22289. Accessed June 24, 2026.
Nampoothiri G, Madhavan, , Prasada Chowdari Gurram, Jayesh Mudgal. "Astrocytic Glutamatergic Transmission in Neurodegenerative Disorders" Encyclopedia, https://encyclopedia.pub/entry/22289 (accessed June 24, 2026).
Nampoothiri G, M., , ., Gurram, P.C., & Mudgal, J. (2022, April 26). Astrocytic Glutamatergic Transmission in Neurodegenerative Disorders. In Encyclopedia. https://encyclopedia.pub/entry/22289
Nampoothiri G, Madhavan, et al. "Astrocytic Glutamatergic Transmission in Neurodegenerative Disorders." Encyclopedia. Web. 26 April, 2022.
Copy Citation
Several neurodegenerative disorders involve impaired neurotransmission, and glutamatergic neurotransmission sets a prototypical example. Glutamate is a predominant excitatory neurotransmitter where the astrocytes play a pivotal role in maintaining the extracellular levels through release and uptake mechanisms. Astrocytes modulate calcium-mediated excitability and release several neurotransmitters and neuromodulators, including glutamate, and significantly modulate neurotransmission. Accumulating evidence supports the concept of excitotoxicity caused by astrocytic glutamatergic release in pathological conditions.
astrocyte
glutamate
neurodegenerative diseases
glia
1. Calcium Mediated Exocytosis
Astrocytes are essential in modulating neuronal activity and synaptic neurotransmission [1]. Nerve terminals are encased with astrocytes and are strategically located to communicate effectively with synapses [1]. The astrocytic responses towards synaptic stimulation have been well established [2]. They mediate Ca2+ dependent glutamate release and regulate synaptic neurotransmission [3]. As the intracellular Ca2+ level required for astrocytic glutamate release is within physiological limits, this release can be exploited as a signaling mechanism to alter synaptic neurotransmission and plasticity within the CNS [4]. Primarily, the generation of intercellular Ca2+ waves (ICW) involves the release of Ca2+ from the ER via G protein coupled receptor (GPCR) activation [5]. Studies on glia revealed the presence of mGluRs which, upon activation by physiological ligands, resulted in the synthesis of inositol 1,4,5-triphosphate (IP3) and subsequent Ca2+ release [6]. This release of Ca2+ promoted the onset and maintenance of ICW of the glial cell, which provided long-range signaling [7]. These waves depict the rise in Ca2+ levels in the cytoplasm, which communicates with other cells and has a wave-like appearance that radiates from its originating source. The ICW is initiated by the release of ATP that follows after hemichannel opening [8]. Interestingly, it was demonstrated that glutamate concentration was directly proportional to the frequency oscillations of Ca2+ waves [9]. This Ca2+ is released by the hippocampal astrocytic cells from intracellular storage both naturally and in response to the activation of Gq-linked GPCR by binding IP3 to its receptor (IP3R). The released Ca2+ in astrocytes is essential and sufficient for the secretion of gliotransmitters, such as ATP and glutamate, and further affects the neuronal activity. IP3R type 2 (IP3R2) appears to be the major IP3R expressed by astrocytes [10]. IP3R-mediated Ca2+ signaling is speculated to cause the activity-dependent and selective release of chemical transmitters. In astrocytes, IP3R2 was once the only known Ca2+ channel; however, Ca2+ imaging techniques have recently determined new Ca2+ sources including mitochondria [11]. Rakers et al. reported that the release of IP3R2-dependent Ca2+ from internal reserves causes a rise in astroglial Ca2+ during neurological disorders including stroke [12]. Astrocytes emit several signaling chemicals such as glutamate, D-serine and ATP. Activities of these molecules, such as modulating synaptic transmission and influencing particular behavior, are vigorously studied, but the identity of their cellular compartments remains unknown [13]. The pharmacological inhibition of vesicular glutamate transporters (VGLUTs) significantly decreased exocytotic glutamate release from astrocytes which is a Ca2+ dependent phenomenon, indicating that these transporters may be instrumental in the astrocytic glutamate release in CNS. VGLUTs transfer cytoplasmic glutamate into exocytotic vesicles, which are propelled by a proton gradient created by vacuolar ATPases (V-ATPases) [14]. VGLUT1 and 2 are also seen, along with synaptic-like vesicles [15]. Montana et al. demonstrated that VGLUTs 1 and 2 are present in rat astrocytes and show high immunoreactivity, justifying their role in the glutamate release via exocytosis [16]. Conversely, the VGLUT mRNAs were absent in astrocytic transcriptome [17]. This supported the notion made by Li et al. that VGLUTs were absent in the astrocytes [18]. The cytosol of astrocytes have high glutamate concentrations ranging from 0.1–5 mM, but extracellular glutamate levels lie within the sub-micromolar range [19]. In astrocytes, glutamate is packed into synaptic-like vesicles and is released in a Ca2+ dependent mechanism, demonstrating the contribution of astrocytes in glutamatergic transmission.
Astrocytes exert controlled glutamate exocytosis via a protein complex called the soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein (SNAP) receptor (SNARE) complex, which regulates vesicle fusion [20]. Synaptobrevin 2, Syntaxin 1, and synaptosome-associated protein of 23 kDa are all part of the core SNARE complex (SNAP-23), while Synaptotagmin 4 is a Ca2+ sensor. SNARE proteins are found on both the vesicular membranes and the presynaptic plasma membranes that cause membrane fusion [21]. In the neurons, the vesicle-associated membrane protein 2 (VAMP2) binds to Syntaxin and synaptosomal-associated protein 25 (SNAP25) on the cell membrane to form the SNARE complex. Synaptotagmin 1, a Ca2+ sensor expressed by neurons, detects the Ca2+ rise caused by Ca2+ entry via voltage-gated Ca2+ channels and triggers the fusion of vesicles to the cell membrane, releasing glutamate. VAMP2/VAMP3, Syntaxin and SNAP25/SNAP23 are all expressed by astrocytes with similar functions [22]. However, Bezzi et al. suggested that, instead of VAMPs, astrocytes express cellubrevin—a SNARE complex of astrocytic vesicles [15]. Further, research revealed that astrocytes produce Synaptotagmins 4, 7, and 11, which cause the release of glutamate from vesicles in response to a rise in intracellular Ca2+ levels in similar fashion to neurons. Intracellular Ca2+ levels need to rise in the range of 250 to 350nM to stimulate astrocytic glutamate release [22][23][24]. With an increase in Ca2+ levels, the vesicles fuse with SNARE proteins and undergo Ca2+-mediated exocytosis to release glutamate [25][26] as shown in Figure 1. Increasing Ca2+ concentrations by overstimulation of glutamate receptors could lead to excitotoxicity and neuronal death [27]. Therefore, impairment in Ca2+ signaling could lead to the progression or worsening of neurodegenerative diseases such as AD and PD.
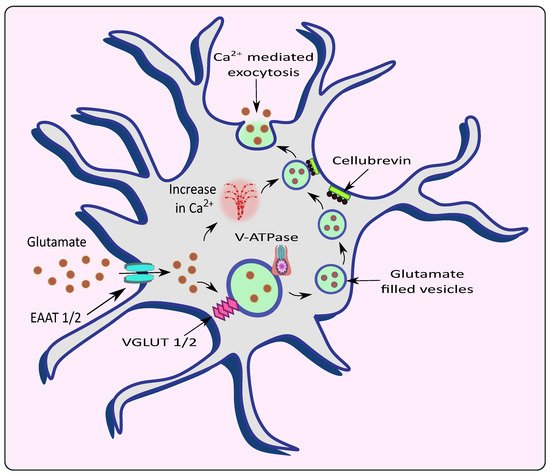
Figure 1. Release of glutamate from astrocytes via Ca2+ mediated exocytosis. Astrocytic EAAT promotes the uptake of glutamate from the synaptic cleft which is filled into the vesicles by VGLUT in the presence of V-ATPase. The rise in intracellular Ca2+ causes the vesicles to fuse with the membrane with the help of an astrocytic vesicular SNARE protein cellubrevin and promotes Ca2+ mediated exocytosis of glutamate.
AD is associated with progressive neurodegeneration and marks its presence primarily through cognitive deficits in patients. Its hallmarks, namely neurofibrillary tangles (NFTs) and the occurrence of amyloid-beta (Aβ) plaques, have been blamed for the progression and worsening of AD [28]. Mostly, it is believed that AD pathology occurs through the amyloid cascade hypothesis originating via the amyloid precursor protein (APP). The action of enzymes namely β secretases and γ secretases produce insoluble Aβ that confers neurotoxicity [29]. Epigenetic modification, proteolysis abnormalities, oxidative stress, neuroinflammation, hampered mitochondrial function, and faulty autophagy are some of the variables that contribute to accelerated aging and neurodegenerative disorders [30][31][32][33]. However, in recent years, the pathology of AD has expanded in multiple dimensions including the pathogenic role of dysfunctional glial cells and the excessive release of neurotransmitters such as glutamate [34].
Neurotoxicity occurs due to an over-accumulation of extracellular glutamate during Aβ aggregation [35][36]. The effect of Aβ 1–42 on a α7 subunit containing nicotinic receptors (7nAChR) could also increase internal Ca2+ currents and subsequent glutamate uptake/release causing glutamate excitotoxicity as shown in Figure 2 [37]. Similarly, Aβ 1–42 has picomolar affinity for the 7nAChR, which is known to enhance glutamate release when activated [38][39]. Lower levels of endogenous Aβ 1–42 are necessary for normal brain function, while at a higher concentration the resultant accumulation and aggregation results in neurotoxicity [40] Through the (7nAChR), Aβ 1–42 can cause glutamate release in the hippocampal nucleus, which is cleared from the extracellular space quickly (msec) by high-affinity EAATs [41][42]. Nicotine-induced glutamate release via the 7nAChR is supported by the hypothesis that Aβ 1–42 binding near the nicotinic site on the 7nAChR can elicit glutamate release [39]. Furthermore, increased Aβ 1–42 synthesis proportionately increases glutamate release in the Cornu Ammonis (CA) 1 area of the amyloid precursor protein/Presenilin 1 (APP/PS1) mouse model [43]. Lower density of 7nAChRs in the CA3 region could explain why an elevated Aβ 1–42 concentration is required to elicit greater glutamate release in the hippocampal region. Aβ 1–42 protein deposition has been reported to occur initially in the CA1 and DG, followed by the CA3 in patients with AD [44]. Hascup and colleagues discovered that enhanced Aβ 1–42 evoked glutamate release in the CA1 and DG at lower doses [45]. The presence of Aβ causes the activation of 7nAChRs present in the astrocytes of the hippocampal regions [46]. Similarly, 7nAChR over expression was observed in the rat astrocytes in the presence of AD pathogenesis [47]. It is evident that 7nAChRs elevate the intracellular Ca2+ levels by stimulating Ca2+ release from intracellular reserves of astrocytes [48].
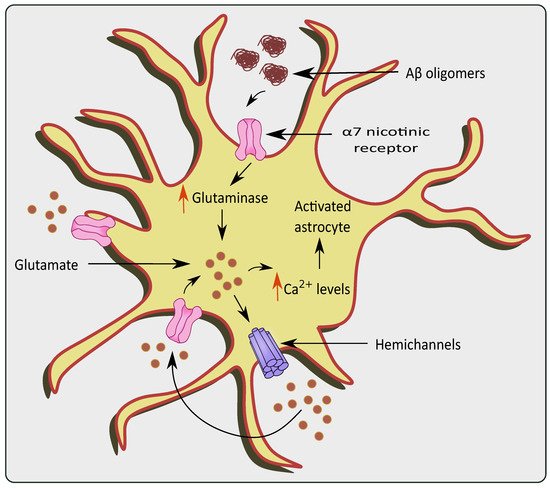
Figure 2. Glutamate excitotoxicity via overstimulation of α 7 nicotinic receptors in the presence of AD pathologies. The presence of Aβ oligomers causes activation and over-stimulation of α 7nAChRs, which increase levels of glutaminase and glutamine in the astrocytes. This causes a rise in Ca2+ levels, ultimately stimulating hemichannels to release more glutamate causing glutamate excitotoxicity.
Similarly, the actions of Aβ 25–35 on the astrocytic purinergic receptors promote Ca2+ level elevation [49]. Chronic calciumopathy, observed in AD, affects neuronal Ca2+ homeostasis and Ca2+ signaling [50]. In AD, the senile plaques promote Ca2+ hyperactivity in astrocytes that trigger excessive glutamate release. The released Ca2+ from the endoplasmic reticulum is essential for astrogliotic response. Therefore this higher Ca2+ signaling, as seen in the entorhinal region and prefrontal cortices of rodent AD models, suggests the abnormalities associated with these pathologies [51]. Additionally, enhanced astroglial Ca2+ signaling followed by glutamate excitotoxicity has been seen in mice with the APP/PS1 gene [52][53]. Recently, Pham et al. found that astrocytic Aβ exposure resulted in both Ca2+ dependent and independent glutamate release, which caused excitotoxicity. Interestingly, a notable amount of glutamate was released before the Ca2+ elevation during Aβ administration, followed by a surge in glutamate with a subsequent rise in Ca2+ [49].
PD is another neurodegenerative disease with impairment in neurons present in the dopaminergic system [54]. The region of substantia nigra pars compacta (SN) residing within the midbrain is highly affected. Pathological hallmarks such as Lewy bodies and α synuclein deposition are considered to play a role in the progression of PD [55]. Patients diagnosed with PD mainly show tremors, muscle rigidity, motor deficits, gait instability, and memory deficits [56]. Recently, autonomic dysfunction has been attributed to PD pathology [57]. Impaired homeostasis of neurotransmitters such as glutamate also plays a pivotal role in the pathogenesis of PD [58]. Inflammatory processes-induced astrocytic glutamate excitotoxicity has also been linked to PD, which causes changes in glutamate transporters and receptor expression [59][60]. Moreover, the accumulation of α-synuclein increases the Ca2+ depolarization-dependent release of presynaptic glutamate as demonstrated using synaptoneurosomes obtained from the forebrain [61]. The α-synuclein releases glutamate in a Ca2+ dependent mechanism, which further activates the extrasynaptic NMDA receptors, and causes neuronal damage [62]. This increase in glutamate activated the AMPA receptors that further upregulate the glutamate release [63]. Interestingly, α-synuclein mobilizes the glutamate vesicles from the pool of reserves [64]. mGluR5 overexcitation upon the binding of α-synuclein also stimulates the release of Ca2+ leading to glutamate excitotoxicity as shown in Figure 3 [65]. A small protein, DJ-1 encoded by PARK7 gene has also been associated with PD pathogenesis, and knockout of PARK7 hampered glutamate uptake via astrocytes. This altered glutamate uptake was associated with the downregulation of EAATs, which led to neurotoxicity in PD patients [66]. Similarly, Wang et al. showed that JWA knockout mice in pathologies of PD reduced the glutamate uptake by hampering EAAT 2 expression [67].
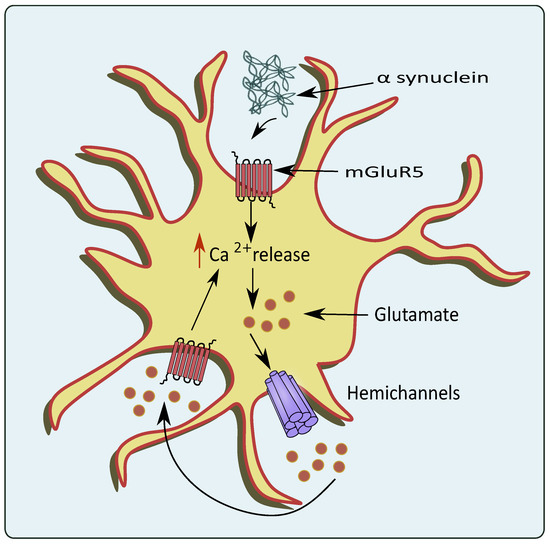
Figure 3. Glutamate excitotoxicity mediated through overexpression of mGluR5 by PD pathologies. In the conditions of PD, the α-synuclein activates astrocytic mGluR5 that elevates intracellular Ca2+ levels and stimulates the release of glutamate through the hemichannels.
ALS is a neurodegenerative disease that involves the degeneration of motor neurons in the CNS [68]. Its prominent characteristic features include motor weakness and loss of motor neurons, gliosis and atrophy of skeletal muscles [69]. Significantly reduced expression of GLT-1 in the motor cortex and spinal cord has been proposed to be one of the main factors leading to glutamate excitotoxicity in ALS [70].
Metadata analysis of studies of transgenic ALS cell cultures suggests that extrinsically boosting the astrocytic GLT-1 level before ALS end-stage may improve the reuptake of glutamate and could be considered a therapeutic strategy. Excitotoxicity could be perhaps due to the ability of those ALS astrocytes that render themselves susceptible to even minor alterations in the neuronal environment. Further, at pre-onset, lowering astrocytic GluR1 levels could help to lessen intracellular Ca2+ [71]. Mutations in valocin containing protein (VCP) genes are causative for ALS. VCP mutant astrocytes showed reduced glutamate uptake and induced reactive astrocytes. This could serve as a protective mechanism at first but becomes toxic over time due to impaired homeostasis. These ALS astrocytes up-regulate inflammatory signaling and are seen to reduce its supportive actions to neurons [72].
In a mouse model of ALS, an increased influx of Ca2+ has been shown to be instrumental in glutamate exocytosis by affecting vesicle fusion and release mechanisms [73]. Furthermore, SOD1 gene mutations in familial ALS interfere with mitochondrial function and prevent glutamate reuptake, thus causing glutamate excitotoxicity [8]. Another interesting target seen in familial ALS is the upregulation of the ATP- binding cassette transporters (ABC) transporter glycoprotein (P-gp). It is known to be upregulated by NMDA receptors activated by glutamate that is excessively secreted by astrocytes with mutant superoxide dismutase 1 (SOD1) [74]. The sporadic and familial mice ALS models have shown a marked reduction in the GLT-1 [75]. Astrocytes that express mutated SOD1 fail to regulate the expression of glutamate receptor’s GluR2 subunit, and is present in motor neurons which leads to higher Ca2+ levels and motor death [76].
Parpura et al. via flash photolysis increased internal Ca2+ in astrocytes to monitor Ca2+ and glutamate levels that elicited slow inward currents. These electrophysiologically recorded signals showed that small variations in astrocytic Ca2+, from 84 nM to 140 nM, elicited large glutamatergic currents in adjacent neurons. Therefore, the astrocytic glutamate release pathway is activated at normal levels of internal Ca2+, as glutamate further elevates Ca2+ in astrocytes to values surpassing 1.8 μM [23]. When cytosolic Ca2+ levels rise, mitochondria quickly absorb Ca2+ to avoid Ca2+ overload in the cytosol. But this excessive mitochondrial Ca2+ uptake could lead to mitochondrial Ca2+ overload and result in events like increase in reactive oxidative species (ROS), glutamate production inhibition of ATP synthesis mitochondrial permeability transition pore (mPTP) opening, the release of cytochrome C, activation of caspases, and apoptosis. The rise in Ca2+ levels is transient in nature and occurs in the presence of disease pathologies involved in neurodegenerative diseases such as AD [23][77], PD [78], multiple sclerosis [79] and Huntington’s disease [80].
Therefore, in such pathological conditions, astrocytes respond rapidly with numerous cellular adaptations, including morphological and functional rearrangements, gene and protein expression alterations, as well as changes in its secretome, which are collectively called reactive astrogliosis [81]. The hallmark features of these reactive astrocytes are cell hypertrophy and up-regulation of Glial fibrillary acidic protein (GFAP) and vimentin (intermediate filaments). Along with these, they exhibit aberrant Ca2+ signaling in a spatial and temporal dependent fashion, depending on the pathological condition [82]. Since these Ca2+-induced events are linked with glutamate exocytosis mediated by Ca2+, their causal role in these types of neurodegenerative disorders needs to be emphasized.
2. Vacuolar ATPases (V-ATPases)
The V-ATPases are proton pumps with multiple subunits composed of a peripheral component V1 attached to an internal membrane-associated component V0 [83]. The V0 is associated with the translocation of proteins while V1 is responsible for the hydrolysis of ATP [84]. These pumps operate in conjunction with eight subunits that are present in V1 and six subunits present in V0 [85]. The regulation of these pumps occurs in multiple processes such as the reversible dissociation of V1 and V0 subunits, disulfide bond formation, the altered proton transport to ATP hydrolysis ratio expressed as coupling efficiency, and modulation in the conductance of counterions [86].
The presence of V-ATPases has been previously identified on the plasma membrane of astrocytes [87] and presynaptic neurons; along with their vesicular membranes [88]. V-ATPases play a role in maintaining acidic pH and the membrane potential to drive the filling of the vesicles with neurotransmitters as shown in Figure 4 [89]. They are also reported to exist in G1 and G2 isoforms where the G2 isoform is instrumental in maintaining the acidification of synaptic vesicles [90]. The Voa1 and Voa2 components of V-ATPases were identified on the vesicles in PC12 cell lines [91]. Interestingly, V-ATPases were recently known to have no direct contribution to the fusion of synaptic vesicles. They are released as V0, V1, and V1C1 components from the acidified vesicles [92]. The V1 deficient synaptic vesicles bind to the plasma membrane and cause the recruitment of other components upon contact with luminal pH. This leads to endocytosis of vesicles that contain the fully assembled V-ATPases [93]. This is followed by the recruitment of H+ ions into the vesicular lumen, the generation of membrane potential, and the filling of glutamate [94].
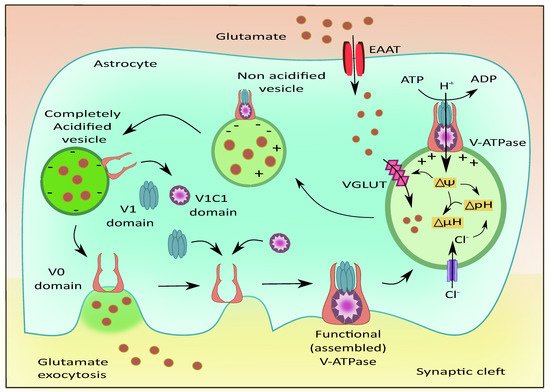
Figure 4. Role of V-ATPases in vesicular glutamate filling and its exocytosis. The subunits of V-ATPases namely V0, V1, and V1C1 assemble to form a functional entity that generates the membrane potential and pH gradient necessary for the uptake of glutamate into the vesicles via VGLUT. Upon complete acidification of the vesicle, the V1 and V1C1 domains detach from the complex while the V0 domain facilitates membrane binding of the vesicle followed by glutamate exocytosis.
The VGLUTs are responsible for recruiting glutamate molecules into the synaptic vesicles [95]. Montana et al. have reported the presence of VGLUTs in astrocytes and their role in astrocytic glutamate transmission [16]. The V-ATPases were localized on the surface of astrocytes present in the hippocampus, and these pumps were responsible for the regulation of intracellular pH (pHi) [96]. The primary astrocytic cultures are mainly dependent on HCO3—independent mechanisms for maintaining the pHi in comparison to cultured astrocytes [97]. The process of neurotransmitter vesicular refilling mainly relies on the electrochemical gradient. This gradient is fulfilled by the V-ATPases and also by chloride ion channels by generating the required chemical gradient and membrane potential to facilitate this uptake [98]. To a small extent, glutamate uptake is dependent on the chemical gradient, while optimum membrane potential is extremely necessary for the vesicular entry of glutamate. Interestingly, since glutamate itself is anionic, it acidifies the vesicles and activates the V-ATPases, enhancing the vesicular filling of glutamate via VGLUT [99]. Another important factor for the optimum functioning of the V-ATPases is the ratio of ADP and ATP in astrocytes. Altered levels of ATP could affect the functioning of these pumps and prevent the development of the required proton gradient that would affect glutamate uptake into the vesicles thus affecting the release of glutamate [100]. The mode of ATP release could be through a Ca2+ dependent method—where the ATP is transported to the plasma membrane of astrocytes through secretory vesicles—and a Ca2+ independent method—where astrocytic hemichannels and purinergic receptors, could release ATP [101].
The blockade of astrocytic V-ATPase with bafilomycin A1, a V-ATPase inhibitor, showed alterations in the expression of TNF-α [102] and low levels of glutamate release in astrocytes [16]. Similarly, the administration of N-ethylmaleimide (NEM) reduced the pHi and incorporation of a more selective inhibitor of V-ATPases, 7-chloro-4-nitroben-2-oxa-1,3-diazole confirmed the vacuolar nature of these pumps [103]. NEM appears to activate potassium ion antiport [104] but the exact mechanism by which it reduces pHi is not known.
The lysosomal degradation is executed by the hydrolytic enzymes, which are activated in acidic pH. The V-ATPase maintains this acidic pH by pumping protons into the lumen, by utilizing ATP [13]. Lysosomal exocytotic release occurs much more slowly as compared to the release of neurotransmitters through vesicles [105]. As the lysosomes contain ATP, lysosomal impairment prevents ATP-mediated Ca2+ release, ultimately affecting astrocytic exocytosis governed by Ca2+ stimulation [106]. This provides evidence for the importance of lysosomal functioning in astrocytes.
The V-ATPases have been implicated in the pathogenesis of neurodegenerative diseases like AD and PD. As discussed earlier, the reversible translocation of V0 and V1 subunits are essential in the optimal functioning of the V-ATPases. The ATP6V1-A was found to be downregulated in the conditions of AD that impacted the neurotransmitter release from synaptic vesicles and prevented phosphorylation and phagosome formation thus worsening AD pathologies [107]. ATP6AP2 is an important accessory protein that promotes neuronal growth in the CNS. Recently, splice variants of ATP6AP2 demonstrated defects in the acidification of lysosomes and progressed towards neuronal death. In in vitro systems, the ATP6AP2 deficits led to impaired V-ATPase assembly, thus affecting its function. The loss or mutations in Presenilin1 (PS1) have contributed to the pathologies of AD [108]. This could be the reason why PS1 deletions showed impaired acidification of lysosomes and impaired autophagy, thus hampering the clearance of oligomers in AD [109].
In the conditions of PD, lysosomal clearance of aggregates, such as α-synuclein, proteins with misfolded morphology, or debris, is essential which is regulated by V-ATPase activity [110]. The mutations in the ATP6AP2 were correlated with progression towards parkinsonism in patients with X-linked parkinsonian syndrome [111]. These studies demonstrate the importance of V-ATPases and their putative implication in neurodegenerative diseases, which is still underexplored.
References
- Ahtiainen, A.; Genocchi, B.; Tanskanen, J.M.A.; Barros, M.T.; Hyttinen, J.A.K.; Lenk, K. Astrocytes exhibit a protective role in neuronal firing patterns under chemically induced seizures in neuron–astrocyte co-cultures. Int. J. Mol. Sci. 2021, 22, 12770.
- Dani, J.W.; Chernjavsky, A.; Smith, S.J. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron 1992, 8, 429–440.
- Hirai, H. Ca2+-dependent regulation of synaptic δ2 glutamate receptor density in cultured rat Purkinje neurons. Eur. J. Neurosci. 2001, 14, 73–82.
- Mielnicka, A.; Michaluk, P. Exocytosis in astrocytes. Biomolecules 2021, 11, 1367.
- Golovina, V.A.; Blaustein, M.P. Unloading and refilling of two classes of spatially resolved endoplasmic reticulum Ca2+ stores in astrocytes. Glia 2000, 31, 15–28.
- Verkhratsky, A.; Kettenmann, H. Calcium signalling in glial cells. Trends Neurosci. 1996, 19, 346–352.
- Parpura, V.; Grubišic, V.; Verkhratsky, A. Ca2+ sources for the exocytotic release of glutamate from astrocytes. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 984–991.
- Grosskreutz, J.; Van Den Bosch, L.; Keller, B.U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 2010, 47, 165–174.
- Cornell-Bell, A.H.; Finkbeiner, S.M.; Cooper, M.S.; Smith, S.J. Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science 1990, 247, 470–473.
- Petravicz, J.; Fiacco, T.A.; McCarthy, K.D. Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J. Neurosci. 2008, 28, 4967–4973.
- Agarwal, A.; Wu, P.H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 2017, 93, 587–605.e7.
- Rakers, C.; Petzold, G.C. Astrocytic calcium release mediates peri-infarct depolarizations in a rodent stroke model. J. Clin. Investig. 2017, 127, 511–516.
- Liu, T.; Sun, L.; Xiong, Y.; Shang, S.; Guo, N.; Teng, S.; Wang, Y.; Liu, B.; Wang, C.; Wang, L.; et al. Calcium Triggers Exocytosis from Two Types of Organelles in a Single Astrocyte. J. Neurosci. 2011, 31, 10593–10601.
- Takamori, S.; Rhee, J.S.; Rosenmund, C.; Jahn, R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 2000, 407, 189–194.
- Bezzi, P.; Gundersen, V.; Galbete, J.L.; Seifert, G.; Steinhäuser, C.; Pilati, E.; Volterra, A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat. Neurosci. 2004, 7, 613–620.
- Montana, V.; Ni, Y.; Sunjara, V.; Hua, X.; Parpura, V. Vesicular Glutamate Transporter-Dependent Glutamate Release from Astrocytes. J. Neurosci. 2004, 24, 2633–2642.
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278.
- Li, D.; Hérault, K.; Silm, K.; Evrard, A.; Wojcik, S.; Oheim, M.; Herzog, E.; Ropert, N. Lack of evidence for vesicular glutamate transporter expression in mouse astrocytes. J. Neurosci. 2013, 33, 4434–4455.
- Cavelier, P.; Attwell, D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J. Physiol. 2005, 564, 397–410.
- Parpura, V.; Fang, Y.; Basarsky, T.; Jahn, R.; Haydon, P.G. Expression of synaptobrevin II, cellubrevin and syntaxin but not SNAP-25 in cultured astrocytes. FEBS Lett. 1995, 377, 489–492.
- Malarkey, E.B.; Parpura, V. Mechanisms of glutamate release from astrocytes. Neurochem. Int. 2008, 52, 142–154.
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184.
- Parpura, V.; Haydon, P.G. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc. Natl. Acad. Sci. USA 2000, 97, 8629–8634.
- Sugita, S.; Han, W.; Butz, S.; Liu, X.; Fernández-Chacón, R.; Lao, Y.; Südhof, T.C. Synaptotagmin VII as a plasma membrane Ca2+ sensor in exocytosis. Neuron 2001, 30, 459–473.
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Amrani, A.; Gris, D. NLRX1 Enhances Glutamate Uptake and Inhibits Glutamate Release by Astrocytes. Cells 2019, 8, 400.
- Zhang, Q.; Pangršič, T.; Kreft, M.; Kržan, M.; Li, N.; Sul, J.Y.; Halassa, M.; Van Bockstaele, E.; Zorec, R.; Haydon, P.G. Fusion-related Release of Glutamate from Astrocytes. J. Biol. Chem. 2004, 279, 12724–12733.
- Albano, R.; Liu, X.Q.; Lobner, D. Regulation of system xc- in the SOD1-G93A mouse model of ALS. Exp. Neurol. 2013, 250, 69–73.
- Lopez, J.A.S.; González, H.M.; Léger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255.
- Uddin, M.S.; Kabir, M.T.; Rahman, M.S.; Behl, T.; Jeandet, P.; Ashraf, G.M.; Najda, A.; Bin-Jumah, M.N.; El-Seedi, H.R.; Abdel-Daim, M.M.; et al. Revisiting the amyloid cascade hypothesis: From anti-aβ therapeutics to auspicious new ways for alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 5858.
- Fernandes, J.; Mudgal, J.; Rao, C.M.; Arora, D.; Mallik, S.B.; Pai, K.S.R.; Nampoothiri, M. N-acetyl-L-tryptophan, a substance-P receptor antagonist attenuates aluminum-induced spatial memory deficit in rats. Toxicol. Mech. Methods 2018, 28, 328–334.
- Ghosh, I.; Sankhe, R.; Mudgal, J.; Arora, D.; Nampoothiri, M. Spermidine, an autophagy inducer, as a therapeutic strategy in neurological disorders. Neuropeptides 2020, 83, 102083.
- Satarker, S.; Maity, S.; Mudgal, J.; Nampoothiri, M. In silico screening of neurokinin receptor antagonists as a therapeutic strategy for neuroinflammation in Alzheimer’s disease. Mol. Divers. 2021, 26, 443–466.
- Balaji, E.V.; Kumar, N.; Satarker, S.; Nampoothiri, M. Zinc as a plausible epigenetic modulator of glioblastoma multiforme. Eur. J. Pharmacol. 2020, 887, 173549.
- Dzamba, D.; Harantova, L.; Butenko, O.; Anderova, M. Glial Cells—The Key Elements of Alzheimers Disease. Curr. Alzheimer Res. 2016, 13, 894–911.
- Jacob, C.P.; Koutsilieri, E.; Bartl, J.; Neuen-Jacob, E.; Arzberger, T.; Zander, N.; Ravid, R.; Roggendorf, W.; Riederer, P.; Grünblatt, E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J. Alzheimers Dis. 2007, 11, 97–116.
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18.
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.I.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527.
- Wang, H.Y.; Lee, D.H.S.; Davis, C.B.; Shank, R.P. Amyloid peptide Aβ1–42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J. Neurochem. 2000, 75, 1155–1161.
- Konradsson-Geuken, A.; Gash, C.R.; Alexander, K.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A.; Bruno, J.P. Second-by-second analysis of alpha 7 nicotine receptor regulation of glutamate release in the prefrontal cortex of awake rats. Synapse 2009, 63, 1069–1082.
- Mura, E.; Lanni, C.; Preda, S.; Pistoia, F.; Sara, M.; Racchi, M.; Schettini, G.; Marchi, M.; Govoni, S. β-Amyloid: A Disease Target or a Synaptic Regulator Affecting Age-Related Neurotransmitter Changes? Curr. Pharm. Des. 2010, 16, 672–683.
- Zhou, Y.; Danbolt, N.C. GABA and glutamate transporters in brain. Front. Endocrinol. 2013, 4, 165.
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105.
- Hascup, K.N.; Hascup, E.R. Altered neurotransmission prior to cognitive decline in AβPP/PS1 mice, a model of Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 771–776.
- Thal, D.R.; Rüb, U.; Schultz, C.; Sassin, I.; Ghebremedhin, E.; Del Tredici, K.; Braak, E.; Braak, H. Sequence of Aβ-protein deposition in the human medial temporal lobe. J. Neuropathol. Exp. Neurol. 2000, 59, 733–748.
- Hascup, K.N.; Hascup, E.R. Soluble Amyloid-β42 Stimulates Glutamate Release through Activation of the α7 Nicotinic Acetylcholine Receptor. J. Alzheimers Dis. 2016, 53, 337–347.
- Pirttimaki, T.M.; Codadu, N.K.; Awni, A.; Pratik, P.; Nagel, D.A.; Hill, E.J.; Dineley, K.T.; Parri, H.R. α7 nicotinic receptor-mediated astrocytic gliotransmitter release: Aβ effects in a preclinical Alzheimer’s mouse model. PLoS ONE 2013, 8, e81828.
- Xiu, J.; Nordberg, A.; Zhang, J.T.; Guan, Z.Z. Expression of nicotinic receptors on primary cultures of rat astrocytes and up-regulation of the α7, α4 and β2 subunits in response to nanomolar concentrations of the β-amyloid peptide1–42. Neurochem. Int. 2005, 47, 281–290.
- Sharma, G.; Vijayaraghavan, S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc. Natl. Acad. Sci. USA 2001, 98, 4148–4153.
- Pham, C.; Hérault, K.; Oheim, M.; Maldera, S.; Vialou, V.; Cauli, B.; Li, D. Astrocytes respond to a neurotoxic Aβ fragment with state-dependent Ca2+ alteration and multiphasic transmitter release. Acta Neuropathol. Commun. 2021, 9, 44.
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic reticulum Ca2+ handling in excitable cells in health and disease. Pharmacol. Rev. 2011, 63, 700–727.
- Verkhratsky, A. Astroglial Calcium Signaling in Aging and Alzheimer’s Disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a035188.
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215.
- Moechars, D.; Lorent, K.; Van Leuven, F. Premature death in transgenic mice that overexpress a mutant amyloid precursor protein is preceded by severe neurodegeneration and apoptosis. Neuroscience 1999, 91, 819–830.
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687.
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12.
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90.
- Chen, Z.; Li, G.; Liu, J. Autonomic dysfunction in Parkinson’s disease: Implications for pathophysiology, diagnosis, and treatment. Neurobiol. Dis. 2020, 134, 104700.
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-induced excitotoxicity in Parkinson’s disease: The role of glial cells. J. Pharmacol. Sci. 2020, 144, 151–164.
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215.
- Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623.
- Sarafian, T.A.; Littlejohn, K.; Yuan, S.; Fernandez, C.; Cilluffo, M.; Koo, B.-K.; Whitelegge, J.P.; Watson, J.B. Stimulation of synaptoneurosome glutamate release by monomeric and fibrillated α-synuclein. J. Neurosci. Res. 2017, 95, 1871–1887.
- Trudler, D.; Sanz-Blasco, S.; Eisele, Y.S.; Ghatak, S.; Bodhinathan, K.; Akhtar, M.W.; Lynch, W.P.; Piña-Crespo, J.C.; Talantova, M.; Kelly, J.W.; et al. A-Synuclein oligomers induce glutamate release from astrocytes and excessive extrasynaptic nmdar activity in neurons, thus contributing to synapse loss. J. Neurosci. 2021, 41, 2264–2273.
- Hüls, S.; Högen, T.; Vassallo, N.; Danzer, K.M.; Hengerer, B.; Giese, A.; Herms, J. AMPA-receptor-mediated excitatory synaptic transmission is enhanced by iron-induced α-synuclein oligomers. J. Neurochem. 2011, 117, 868–878.
- Gureviciene, I.; Gurevicius, K.; Tanila, H. Role of α-synuclein in synaptic glutamate release. Neurobiol. Dis. 2007, 28, 83–89.
- Price, D.L.; Rockenstein, E.; Ubhi, K.; Phung, V.; Maclean-Lewis, N.; Askay, D.; Cartier, A.; Spencer, B.; Patrick, C.; Desplats, P.; et al. Alterations in mGluR5 expression and signaling in lewy body disease and in transgenic models of alpha- synucleinopathy—Implications for excitotoxicity. PLoS ONE 2010, 5, e14020.
- Kim, J.M.; Cha, S.H.; Choi, Y.R.; Jou, I.; Joe, E.H.; Park, S.M. DJ-1 deficiency impairs glutamate uptake into astrocytes via the regulation of flotillin-1 and caveolin-1 expression. Sci. Rep. 2016, 6, 28823.
- Wang, R.; Zhao, X.; Xu, J.; Wen, Y.; Li, A.; Lu, M.; Zhou, J. Astrocytic JWA deletion exacerbates dopaminergic neurodegeneration by decreasing glutamate transporters in mice. Cell Death Dis. 2018, 9, 352.
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700.
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38.
- Vaz, S.H.; Pinto, S.; Sebastião, A.M.; Brites, D. Astrocytes in Amyotrophic Lateral Sclerosis. In Amyotrophic Lateral Sclerosis; Exon Publications: Brisbane, Australia, 2021; pp. 35–54.
- Jordan, K.; Murphy, J.; Singh, A.; Mitchell, C.S. Astrocyte-mediated neuromodulatory regulation in preclinical ALS: A metadata analysis. Front. Cell. Neurosci. 2018, 12, 491.
- Ziff, O.J.; Clarke, B.E.; Taha, D.M.; Crerar, H.; Luscombe, N.M.; Patani, R. Meta-analysis of human and mouse ALS astrocytes reveals multi-omic signatures of inflammatory reactive states. Genome Res. 2022, 32, 71–84.
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal exocytotic release of glutamate in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2011, 116, 1028–1042.
- Mohamed, L.A.; Markandaiah, S.S.; Bonanno, S.; Pasinelli, P.; Trotti, D. Excess glutamate secreted from astrocytes drives upregulation of P-glycoprotein in endothelial cells in amyotrophic lateral sclerosis. Exp. Neurol. 2019, 316, 27–38.
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 2002, 99, 1604–1609.
- Van Damme, P.; Bogaert, E.; Dewil, M.; Hersmus, N.; Kiraly, D.; Scheveneels, W.; Bockx, I.; Braeken, D.; Verpoorten, N.; Verhoeven, K.; et al. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc. Natl. Acad. Sci. USA 2007, 104, 14825–14830.
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146.
- Bantle, C.M.; Hirst, W.D.; Weihofen, A.; Shlevkov, E. Mitochondrial Dysfunction in Astrocytes: A Role in Parkinson’s Disease? Front. Cell Dev. Biol. 2021, 8, 608026.
- Enders, M.; Heider, T.; Ludwig, A.; Kuerten, S. Strategies for neuroprotection in multiple sclerosis and the role of calcium. Int. J. Mol. Sci. 2020, 21, 1663.
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci. 2016, 36, 3453–3470.
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434.
- Shigetomi, E.; Saito, K.; Sano, F.; Koizumi, S.C. Aberrant calcium signals in reactive astrocytes: A key process in neurological disorders. Int. J. Mol. Sci. 2019, 20, 996.
- Kane, P.M. The Where, When, and How of Organelle Acidification by the Yeast Vacuolar H+-ATPase. Microbiol. Mol. Biol. Rev. 2006, 70, 177–191.
- Forgac, M. Structure and properties of the vacuolar (H+)-ATPases. J. Biol. Chem. 1999, 274, 12951–12954.
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929.
- Collins, M.P.; Forgac, M. Regulation and function of V-ATPases in physiology and disease. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183341.
- Philippe, J.M.; Dubois, J.M.; Rouzaire-Dubois, B.; Cartron, P.F.; Vallette, F.; Morel, N. Functional expression of V-ATPases in the plasma membrane of glial cells. Glia 2002, 37, 365–373.
- Morel, N.; Dedieu, J.C.; Philippe, J.M. Specific sorting of the a1 isoform of the V-H+ATPase a subunit to nerve terminals where it associates with both synaptic vesicles and the presynaptic plasma membrane. J. Cell Sci. 2003, 116, 4751–4762.
- Ueda, T. Vesicular Glutamate Uptake. Adv. Neurobiol. 2016, 13, 173–221.
- Murata, Y.; Sun-Wada, G.H.; Yoshimizu, T.; Yamamoto, A.; Wada, Y.; Futai, M. Differential localization of the vacuolar H+ pump with G subunit isoforms (G1 and G2) in mouse neurons. J. Biol. Chem. 2002, 277, 36296–36303.
- Saw, N.M.N.; Kang, S.Y.A.; Parsaud, L.; Han, G.A.; Jiang, T.; Grzegorczyk, K.; Surkont, M.; Sun-Wada, G.-H.; Wada, Y.; Li, L.; et al. Vacuolar H+-ATPase subunits Voa1 and Voa2 cooperatively regulate secretory vesicle acidification, transmitter uptake, and storage. Mol. Biol. Cell 2011, 22, 3394–3409.
- Morel, N.; Poëa-Guyon, S. The membrane domain of vacuolar H+ ATPase: A crucial player in neurotransmitter exocytotic release. Cell. Mol. Life Sci. 2015, 72, 2561–2573.
- Bodzęta, A.; Kahms, M.; Klingauf, J. The Presynaptic v-ATPase Reversibly Disassembles and Thereby Modulates Exocytosis but Is Not Part of the Fusion Machinery. Cell Rep. 2017, 20, 1348–1359.
- Nishi, T.; Forgac, M. The vacuolar (H+)-ATPases—Nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 2002, 3, 94–103.
- Liguz-Lecznar, M.; Skangiel-Kramska, J. Vesicular glutamate transporters (VGLUTs): The three musketeers of glutamatergic system. Acta Neurobiol. Exp. 2007, 67, 207–218.
- Pappas, C.A.; Ransom, B.R. A depolarization-stimulated, bafilomycin-inhibitable H+ pump in hippocampal astrocytes. Glia 1993, 9, 280–291.
- Hansen, D.B.; Garrido-Comas, N.; Salter, M.; Fern, R. HCO3−-independent pH regulation in astrocytes in Situ is dominated by V-ATPase. J. Biol. Chem. 2015, 290, 8039–8047.
- Martineau, M.; Guzman, R.E.; Fahlke, C.; Klingauf, J. VGLUT1 functions as a glutamate/proton exchanger with chloride channel activity in hippocampal glutamatergic synapses. Nat. Commun. 2017, 8, 2279.
- Hnasko, T.S.; Edwards, R.H. Neurotransmitter Corelease: Mechanism and Physiological Role. Annu. Rev. Physiol. 2012, 74, 225–243.
- Reyes, R.C.; Parpura, V. Mitochondria modulate Ca2+-dependent glutamate release from rat cortical astrocytes. J. Neurosci. 2008, 28, 9682–9691.
- Xiong, Y.; Sun, S.; Teng, S.; Jin, M.; Zhou, Z. Ca2+-Dependent and Ca2+-Independent ATP Release in Astrocytes. Front. Mol. Neurosci. 2018, 11, 224.
- Conboy, I.M.; Manoli, D.; Mhaiskar, V.; Jones, P.P. Calcineurin and vacuolar-type H+-ATPase modulate macrophage effector functions. Proc. Natl. Acad. Sci. USA 1999, 96, 6324–6329.
- Volk, C.; Albert, T.; Kempski, O.S. A proton-translocating H+-ATPase is involved in C6 Glial pH regulation. Biochim. Biophys. Acta Biomembr. 1998, 1372, 28–36.
- Elmore, M.J.; Lamb, A.J.; Ritchie, G.Y.; Douglas, R.M.; Munro, A.; Gajewska, A.; Booth, I.R. Activation potassium efflux from Escherichia coli by glutathione metabolites. Mol. Microbiol. 1990, 4, 405–412.
- Li, D.; Ropert, N.; Koulakoff, A.; Giaume, C.; Oheim, M. Lysosomes are the major vesicular compartment undergoing Ca2+-regulated exocytosis from cortical astrocytes. J. Neurosci. 2008, 28, 7648–7658.
- Zhang, Z.; Chen, G.; Zhou, W.; Song, A.; Xu, T.; Luo, Q.; Wang, W.; Gu, X.-S.; Duan, S. Regulated ATP release from astrocytes through lysosome exocytosis. Nat. Cell Biol. 2007, 9, 945–953.
- Zhou, Z.; Bai, J.; Zhong, S.; Zhang, R.; Kang, K.; Zhang, X.; Xu, Y.; Zhao, C.; Zhao, M. Downregulation of ATP6V1A Involved in Alzheimer’s Disease via Synaptic Vesicle Cycle, Phagosome, and Oxidative Phosphorylation. Oxid. Med. Cell. Longev. 2021, 2021, 5555634.
- Deyts, C.; Clutter, M.; Herrera, S.; Jovanovic, N.; Goddi, A.; Parent, A.T. Loss of presenilin function is associated with a selective gain of APP function. eLife 2016, 5, e15645.
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158.
- Song, Q.; Meng, B.; Xu, H.; Mao, Z. The emerging roles of vacuolar-type ATPase-dependent Lysosomal acidification in neurodegenerative diseases. Transl. Neurodegener. 2020, 9, 17.
- Korvatska, O.; Strand, N.S.; Berndt, J.D.; Strovas, T.; Chen, D.H.; Leverenz, J.B.; Kiianitsa, K.; Mata, I.F.; Karakoc, E.; Greenup, J.L.; et al. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum. Mol. Genet. 2013, 22, 3259–3268.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
750
Revisions:
2 times
(View History)
Update Date:
27 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No