 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | MIchèle Studer | -- | 4380 | 2022-04-22 16:03:49 | | | |
| 2 | Amina Yu | + 346 word(s) | 4726 | 2022-04-24 03:57:45 | | | | |
| 3 | Amina Yu | -346 word(s) | 4380 | 2022-04-24 04:02:34 | | | | |
| 4 | Amina Yu | + 347 word(s) | 4727 | 2022-04-24 04:17:30 | | | | |
| 5 | MIchèle Studer | -1 word(s) | 4726 | 2022-04-27 17:56:29 | | |
Video Upload Options
Bosch–Boonstra–Schaaf Optic Atrophy Syndrome (BBSOAS; OMIM 615722; ORPHA 401777) is a genetic neurodevelopmental syndrome caused by the haploinsufficiency of the NR2F1 gene, a key transcriptional regulator of brain and eye development. Although intellectual disability, developmental delay and visual impairment are arguably the most common symptoms affecting BBSOAS patients, multiple additional features are often reported, including epilepsy, autistic traits and hypotonia. These features can be present alone or as comorbidities, with a severity degree that presumably varies depending on the type of NR2F1 genetic perturbation, following a still not well characterized genotype–phenotype correlation. Pathogenic BBSOAS point mutations are principally located in the two most conserved functional domains of the NR2F1 protein: the DNA-binding domain (DBD), responsible for the interaction with target gene regulatory sequences, and the ligand-binding domain (LBD), necessary for dimerization and co-factor binding.
1. Optic Atrophy (OA), Optic Nerve Hypoplasia (ONH) and Optic Disc Abnormalities in Bosch–Boonstra–Schaaf Optic Atrophy Syndrome (BBSOAS) Patients
In light of its several and variable clinical features affecting the visual system, such as OA or optic nerve pallor, ONH or small optic discs, cerebral visual impairment (CVI), nystagmus (uncontrolled eye movements) and alacrima (decreased tear reflex), BBSOAS can also be defined as a congenital optic neuropathy. One of the main and first-reported visual impairments in BBSOAS children is OA, as the name of the syndrome suggests. It can be defined as optic nerve (ON) damage anywhere from the retina to the lateral geniculate nucleus of the thalamus, usually caused by retinal ganglion cell (RGC) death and retraction of RGC axons, with a resulting pale ON in fundoscopy.
However, OA and ONH have both been reported since the first description of BBSOAS patients [1]. As OA, ONH is also characterized by a deficiency of RGCs and their axons, leading to ganglion cell layer disorganization and a small optic disc with a thin ON. A main difference between ONH and OA is that while ONH is a congenital, non-progressive disease characterized by underdevelopment of the ON, OA is instead degenerative with a normal early development of the ON that deteriorates over time. according to experimental data in Nr2f1-deficient mouse models, BBSOAS-like visual impairments, such as retinal and ON anomalies, result from primary RGC patterning, myelination and inflammatory defects originating during early development (Figure 1) [2][3], and leading to visual system defects that would remain stable during postnatal life [3]. Consistently with early appearance of BBSOAS, ONH could be the major clinical feature accounting for the non-progressive visual dysfunction observed in patients [3].
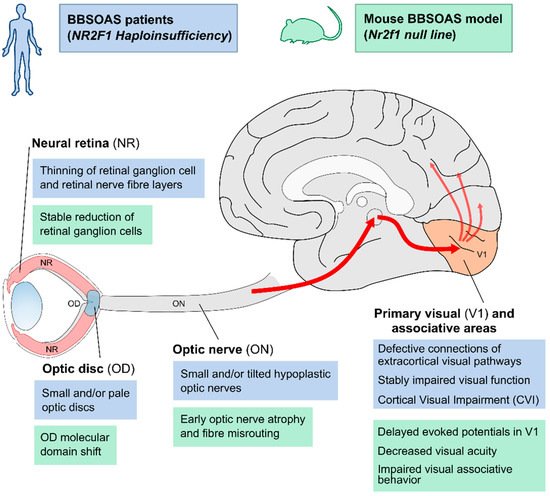
In addition, several BBSOAS patients show small malformations of the optic disc (OD) at the edge between the neural retina and the ON. Such malformations manifest in excavated, pale or small ODs. Often associated with other congenital eye malformations, OD lesions can negatively affect visual acuity. Interestingly, OD malformations are reported more frequently in patients with NR2F1 deletions (33%) than in patients with other variants (12–22%). A structural change in the form of pale and/or excavated ODs is diagnosed by ophthalmologists as an indirect sign of underlying ON diseases, including OA or ONH. In fact, OD pallor and abnormal shape can result from RGC death (causing axonal fibers loss) and degeneration of pial capillaries entering the optic nerve head [4][5][6]. However, some OD malformations—such as optic disc coloboma—can be the direct consequence of genetic or environmental insults affecting the development of this region, rather than a consequence of ON disease [7][8][9]. The OD pit, for example, consisting in a round or oval localized depression within the OD, is caused by defective occlusion of the embryonic ventral fissure of the optic vesicle [10][11]. By homology, data from Nr2f1 mouse models showed delayed ventral fissure closure in null embryos [2] or severe coloboma in double Nr2f1 and Nr2f2 conditional mutants [12], suggesting that impaired ventral fissure fusion could be responsible for OD malformations in a Nr2f1-deficient context. This would also fit with reports of BBSOAS patients showing coloboma-like malformations [13][14][15].
2. When Intellectual Disability Meets Visual Disfunction: Cortical Visual Impairment (CVI) in BBSOAS Patients
| BBSOAS Main Feature(s) | Clinical Description(s) |
|---|---|
| Developmental delay (DD) | Delay in reaching language, social or motor skills milestones |
| Intellectual disability (ID) | Significantly reduced ability to understand new or complex information and to learn and apply new skills (impaired intelligence). IQ ranging from profound ID with IQ < 20, to moderate (35 < IQ < 49) or mild ID (50 < IQ < 69) |
| Visual impairment | Optic nerve abnormalities and/or brain-based vision impairment: |
| Optic nerve atrophy or pallor | |
| Optic nerve hypoplasia | |
| Cortical visual impairment (difficulty locating objects in a crowded field and following rapidly moving images and scenes). | |
| Alacrima (abnormal amount of reflex tearing) | |
| Manifest latent nystagmus and poor tracking; congenital | |
| Significant refractive errors | |
| Amblyopia | |
| Hypotonia | Low muscle tone; congenital |
| Oromotor dysfunction | Swallowing, sucking and chewing problems; congenital |
| Repetitive behavior | Hand flapping, head banging and more |
| Autism spectrum disorder (ASD) | ASD or autistic traits |
| Seizures | Infantile and/or febrile; occipital seizures |
| Attention-deficit hyperactivity disorder (ADHD) | Inattention, impulsivity and hyperactivity |
| Hearing impairment | Abnormal hearing |
| Dysmorphic facial features | Mild and inconsistent |
| Thin corpus callosum and neocortical dysgyria | Hypoplasia of the corpus callosum and abnormal pattern of cortical convolutions and sulci (dysgyria in temporal and parietal areas) on brain MRI |
3. The Many Converging Roads of Intellectual Disability in BBSOAS Patients: From Corpus-Callosum Thinning to Hippocampal and Neocortical Malformations
4. NR2F1 as an Autism Spectrum Disorder Gene
5. NR2F1 as a Susceptibility Gene for Infantile Epileptic Disorders
6. NR2F1 on the Move: Motor Dysfunction in BBSOAS Patients
References
- Bosch, D.G.M.; Boonstra, F.N.; Gonzaga-Jauregui, C.; Xu, M.; de Ligt, J.; Jhangiani, S.; Wiszniewski, W.; Muzny, D.M.; Yntema, H.G.; Pfundt, R.; et al. NR2F1 Mutations Cause Optic Atrophy with Intellectual Disability. Am. J. Hum. Genet. 2014, 94, 303–309.
- Bertacchi, M.; Gruart, A.; Kaimakis, P.; Allet, C.; Serra, L.; Giacobini, P.; Delgado-García, J.M.; Bovolenta, P.; Studer, M. Mouse Nr2f1 Haploinsufficiency Unveils New Pathological Mechanisms of a Human Optic Atrophy Syndrome. EMBO Mol. Med. 2019, 11, e10291.
- Jurkute, N.; Bertacchi, M.; Arno, G.; Tocco, C.; Kim, U.S.; Kruszewski, A.M.; Avery, R.A.; Bedoukian, E.C.; Han, J.; Ahn, S.J.; et al. Pathogenic NR2F1 Variants Cause a Developmental Ocular Phenotype Recapitulated in a Mutant Mouse Model. Brain Commun. 2021, 3, fcab162.
- McClard, C.K.; Shah, V. Pediatric Optic Disc Pallor. Int. Ophthalmol. Clin. 2018, 58, 125–145.
- Jonas, J.B.; Budde, W.M.; Panda-Jonas, S. Ophthalmoscopic Evaluation of the Optic Nerve Head. Surv. Ophthalmol. 1999, 43, 293–320.
- Rodenbeck, S.J.; Mackay, D.D. Examining the Ocular Fundus in Neurology. Curr. Opin. Neurol. 2019, 32, 105–110.
- Cekić, S.; Stanković-Babić, G.; Višnjić, Z.; Jovanović, I.; Risimić, D. Optic Disc Abnormalities—Diagnosis, Evolution and Influence on Visual Acuity. Bosn. J. Basic Med. Sci. 2010, 10, 125–132.
- Dutton, G.N. Congenital Disorders of the Optic Nerve: Excavations and Hypoplasia. Eye 2004, 18, 1038–1048.
- Jeng-Miller, K.W.; Cestari, D.M.; Gaier, E.D. Congenital Anomalies of the Optic Disc: Insights from Optical Coherence Tomography Imaging. Curr. Opin. Ophthalmol. 2017, 28, 579–586.
- Anglo, M. Topical Review: Optic Disc Pits and Associated Maculopathy. Optom. Vis. Sci. 2020, 97, 531–535.
- Uzel, M.M.; Karacorlu, M. Optic Disk Pits and Optic Disk Pit Maculopathy: A Review. Surv. Ophthalmol. 2019, 64, 595–607.
- Tang, K.; Xie, X.; Park, J.-I.; Jamrich, M.; Tsai, S.; Tsai, M.-J. COUP-TFs Regulate Eye Development by Controlling Factors Essential for Optic Vesicle Morphogenesis. Development 2010, 137, 725–734.
- Cardoso, C.; Boys, A.; Parrini, E.; Mignon-Ravix, C.; McMahon, J.M.; Khantane, S.; Bertini, E.; Pallesi, E.; Missirian, C.; Zuffardi, O.; et al. Periventricular Heterotopia, Mental Retardation, and Epilepsy Associated with 5q14.3-Q15 Deletion. Neurology 2009, 72, 784–792.
- Chen, C.-A.; Bosch, D.G.M.; Cho, M.T.; Rosenfeld, J.A.; Shinawi, M.; Lewis, R.A.; Mann, J.; Jayakar, P.; Payne, K.; Walsh, L.; et al. The Expanding Clinical Phenotype of Bosch-Boonstra-Schaaf Optic Atrophy Syndrome: 20 New Cases and Possible Genotype-Phenotype Correlations. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 1143–1150.
- Gazdagh, G.; Mawby, R.; Self, J.E.; Baralle, D. Deciphering Developmental Disorders Study A Severe Case of Bosch– Boonstra–Schaaf Optic Atrophy Syndrome with a Novel Description of Coloboma and Septo-optic Dysplasia, Owing to a Start Codon Variant in the NR2F1 Gene. Am. J. Med. Genet. A 2022, 188, 900–906.
- Tang, K.; Tsai, S.Y.; Tsai, M.-J. COUP-TFs and Eye Development. Biochim. Biophys. Acta 2015, 1849, 201–209.
- Deiner, M.S.; Kennedy, T.E.; Fazeli, A.; Serafini, T.; Tessier-Lavigne, M.; Sretavan, D.W. Netrin-1 and DCC Mediate Axon Guidance Locally at the Optic Disc: Loss of Function Leads to Optic Nerve Hypoplasia. Neuron 1997, 19, 575–589.
- Morcillo, J. Proper Patterning of the Optic Fissure Requires the Sequential Activity of BMP7 and SHH. Development 2006, 133, 3179–3190.
- Chung, G.W.; Edwards, A.O.; SchimentiI, L.A.; Manligas, G.S.; Zhang, Y.; Ritter, R. Renal-Coloboma Syndrome: Report of a Novel PAX2 Gene Mutation. Am. J. Ophthalmol. 2001, 132, 910–914.
- Eccles, M.R.; Schimmenti, L.A. Renal-Coloboma Syndrome: A Multi-System Developmental Disorder Caused by PAX2 Mutations: PAX2 Mutation in Renal-Coloboma Syndrome. Clin. Genet. 1999, 56, 1–9.
- Good, V.; Jan, J.E.; Barrovich, A.J.; Hoyt, S. Cortical Visual Impairment in Children. 14. Surv. Ophthalmol. 1994, 38, 351–364.
- Good, W.V.; Jan, J.E.; Burden, S.K.; Skoczenski, A.; Candy, R. Recent Advances in Cortical Visual Impairment. Dev. Med. Child Neurol. 2007, 43, 56–60.
- Rech, M.E.; McCarthy, J.M.; Chen, C.-A.; Edmond, J.C.; Shah, V.S.; Bosch, D.G.M.; Berry, G.T.; Williams, L.; Madan-Khetarpal, S.; Niyazov, D.; et al. Phenotypic Expansion of Bosch-Boonstra-Schaaf Optic Atrophy Syndrome and Further Evidence for Genotype-Phenotype Correlations. Am. J. Med. Genet. Part A 2020, 182, 1426–1437.
- Hoyt, C.S. Visual Function in the Brain-Damaged Child. Eye 2003, 17, 369–384.
- Huo, R.; Burden, S.K.; Hoyt, C.S.; Good, W.V. Chronic Cortical Visual Impairment in Children: Aetiology, Prognosis, and Associated Neurological Deficits. Br. J. Ophthalmol. 1999, 83, 670–675.
- Jan, J.E.; Groenveld, M.; Sykanda, A.M.; Hoyt, C.S. Behavioural Characteristics Of Children With Permanent Cortical Visual Impairment. Dev. Med. Child Neurol. 2008, 29, 571–576.
- Malkowicz, D.E.; Myers, G.; Leisman, G. Rehabilitation of Cortical Visual Impairment in Children. Int. J. Neurosci. 2006, 116, 1015–1033.
- McConnell, E.L.; Saunders, K.J.; Little, J. What Assessments Are Currently Used to Investigate and Diagnose Cerebral Visual Impairment (CVI) in Children? A Systematic Review. Ophthalmic Physiol. Opt. 2021, 41, 224–244.
- Philip, S.S.; Dutton, G.N. Identifying and Characterising Cerebral Visual Impairment in Children: A Review: Cerebral Visual Impairment in Children: A Review. Clin. Exp. Optom. 2014, 97, 196–208.
- Sakki, H.E.A.; Dale, N.J.; Sargent, J.; Perez-Roche, T.; Bowman, R. Is There Consensus in Defining Childhood Cerebral Visual Impairment? A Systematic Review of Terminology and Definitions. Br. J. Ophthalmol. 2018, 102, 424–432.
- Bojanek, E.K.; Mosconi, M.W.; Guter, S.; Betancur, C.; Macmillan, C.; Cook, E.H. Clinical and Neurocognitive Issues Associated with Bosch-Boonstra-Schaaf Optic Atrophy Syndrome: A Case Study. Am. J. Med. Genet. Part A 2019, 182, 213–218.
- Zhou, C. COUP-TFI: An Intrinsic Factor for Early Regionalization of the Neocortex. Genes Dev. 2001, 15, 2054–2059.
- Armentano, M.; Chou, S.-J.; Srubek Tomassy, G.; Leingärtner, A.; O’Leary, D.D.M.; Studer, M. COUP-TFI Regulates the Balance of Cortical Patterning between Frontal/Motor and Sensory Areas. Nat. Neurosci. 2007, 10, 1277–1286.
- Chou, S.-J.; Babot, Z.; Leingartner, A.; Studer, M.; Nakagawa, Y.; O’Leary, D.D.M. Geniculocortical Input Drives Genetic Distinctions Between Primary and Higher-Order Visual Areas. Science 2013, 340, 1239–1242.
- Cadwell, C.R.; Bhaduri, A.; Mostajo-Radji, M.A.; Keefe, M.G.; Nowakowski, T.J. Development and Arealization of the Cerebral Cortex. Neuron 2019, 103, 980–1004.
- Alfano, C.; Studer, M. Neocortical Arealization: Evolution, Mechanisms, and Open Questions. Dev. Neurobiol. 2013, 73, 411–447.
- Alfano, C. The Nuclear Receptors COUP-TF: A Long-Lasting Experience in Forebrain Assembly. Cell. Mol. Life Sci. 2014, 71, 43–62.
- Flore, G.; Di Ruberto, G.; Parisot, J.; Sannino, S.; Russo, F.; Illingworth, E.A.; Studer, M.; De Leonibus, E. Gradient COUP-TFI Expression Is Required for Functional Organization of the Hippocampal Septo-Temporal Longitudinal Axis. Cereb. Cortex 2016, 27, bhv336.
- Alzu’bi, A.; Lindsay, S.; Kerwin, J.; Looi, S.J.; Khalil, F.; Clowry, G.J. Distinct Cortical and Sub-Cortical Neurogenic Domains for GABAergic Interneuron Precursor Transcription Factors NKX2.1, OLIG2 and COUP-TFII in Early Fetal Human Telencephalon. Brain Struct. Funct. 2017, 222, 2309–2328.
- Foglio, B.; Rossini, L.; Garbelli, R.; Regondi, M.C.; Mercurio, S.; Bertacchi, M.; Avagliano, L.; Bulfamante, G.; Coras, R.; Maiorana, A.; et al. Dynamic Expression of NR2F1 and SOX2 in Developing and Adult Human Cortex: Comparison with Cortical Malformations. Brain Struct. Funct. 2021, 226, 1303–1322.
- Hofman, J.; Hutny, M.; Sztuba, K.; Paprocka, J. Corpus Callosum Agenesis: An Insight into the Etiology and Spectrum of Symptoms. Brain Sci. 2020, 10, 625.
- Paul, L.K.; Brown, W.S.; Adolphs, R.; Tyszka, J.M.; Richards, L.J.; Mukherjee, P.; Sherr, E.H. Agenesis of the Corpus Callosum: Genetic, Developmental and Functional Aspects of Connectivity. Nat. Rev. Neurosci. 2007, 8, 287–299.
- Schell-Apacik, C.C.; Wagner, K.; Bihler, M.; Ertl-Wagner, B.; Heinrich, U.; Klopocki, E.; Kalscheuer, V.M.; Muenke, M.; von Voss, H. Agenesis and Dysgenesis of the Corpus Callosum: Clinical, Genetic and Neuroimaging Findings in a Series of 41 Patients. Am. J. Med. Genet. A 2008, 146A, 2501–2511.
- Alfano, C.; Viola, L.; Heng, J.I.-T.; Pirozzi, M.; Clarkson, M.; Flore, G.; De Maio, A.; Schedl, A.; Guillemot, F.; Studer, M. COUP-TFI Promotes Radial Migration and Proper Morphology of Callosal Projection Neurons by Repressing Rnd2 Expression. Development 2011, 138, 4685–4697.
- Armentano, M.; Filosa, A.; Andolfi, G.; Studer, M. COUP-TFI Is Required for the Formation of Commissural Projections in the Forebrain by Regulating Axonal Growth. Development 2006, 133, 4151–4162.
- Tenembaum, S.N. Acute Disseminated Encephalomyelitis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 112, pp. 1253–1262. ISBN 978-0-444-52910-7.
- Yamaguchi, H.; Zhou, C.; Lin, S.-C.; Durand, B.; Tsai, S.Y.; Tsai, M.-J. The Nuclear Orphan Receptor COUP-TFI Is Important for Differentiation of Oligodendrocytes. Dev. Biol. 2004, 266, 238–251.
- Broadbent, N.J.; Squire, L.R.; Clark, R.E. Spatial Memory, Recognition Memory, and the Hippocampus. Proc. Natl. Acad. Sci. USA 2004, 101, 14515–14520.
- Hannula, D.E.; Libby, L.A.; Yonelinas, A.P.; Ranganath, C. Medial Temporal Lobe Contributions to Cued Retrieval of Items and Contexts. Neuropsychologia 2013, 51, 2322–2332.
- Kumaran, D.; Maguire, E.A. The Human Hippocampus: Cognitive Maps or Relational Memory? J. Neurosci. 2005, 25, 7254–7259.
- Mátéffyová, A.; Otáhal, J.; Tsenov, G.; Mareš, P.; Kubová, H. Intrahippocampal Injection of Endothelin-1 in Immature Rats Results in Neuronal Death, Development of Epilepsy and Behavioral Abnormalities Later in Life. Eur. J. Neurosci. 2006, 24, 351–360.
- Ramos, J.M.J. Hippocampal Damage Impairs Long-Term Spatial Memory in Rats: Comparison between Electrolytic and Neurotoxic Lesions. Physiol. Behav. 2008, 93, 1078–1085.
- Wan, J.; Shen, C.M.; Wang, Y.; Wu, Q.Z.; Wang, Y.L.; Liu, Q.; Sun, Y.M.; Cao, J.P.; Wu, Y.Q. Repeated Exposure to Propofol in the Neonatal Period Impairs Hippocampal Synaptic Plasticity and the Recognition Function of Rats in Adulthood. Brain Res. Bull. 2021, 169, 63–72.
- Zhao, A.; Fang, F.; Li, B.; Chen, Y.; Qiu, Y.; Wu, Y.; Xu, W.; Deng, Y. Visual Abnormalities Associate With Hippocampus in Mild Cognitive Impairment and Early Alzheimer’s Disease. Front. Aging Neurosci. 2021, 12, 522.
- Chen, C.-A.; Wang, W.; Pedersen, S.E.; Raman, A.; Seymour, M.L.; Ruiz, F.R.; Xia, A.; van der Heijden, M.E.; Wang, L.; Yin, J.; et al. Nr2f1 Heterozygous Knockout Mice Recapitulate Neurological Phenotypes of Bosch-Boonstra-Schaaf Optic Atrophy Syndrome and Show Impaired Hippocampal Synaptic Plasticity. Hum. Mol. Genet. 2020, 29, 705–715.
- Parisot, J.; Flore, G.; Bertacchi, M.; Studer, M. COUP-TFI Mitotically Regulates Production and Migration of Dentate Granule Cells and Modulates Hippocampal Cxcr4 Expression. Dev. Camb. Engl. 2017, 144, 2045–2058.
- Moser, M.B.; Moser, E.I.; Forrest, E.; Andersen, P.; Morris, R.G.M. Spatial Learning with a Minislab in the Dorsal Hippocampus. Proc. Natl. Acad. Sci. USA 1995, 92, 9697–9701.
- Bertacchi, M.; Romano, A.L.; Loubat, A.; Tran Mau-Them, F.; Willems, M.; Faivre, L.; Khau van Kien, P.; Perrin, L.; Devillard, F.; Sorlin, A.; et al. NR2F1 Regulates Regional Progenitor Dynamics in the Mouse Neocortex and Cortical Gyrification in BBSOAS Patients. EMBO J. 2020, 39, e104163.
- Oberhuber, M.; Hope, T.M.H.; Seghier, M.L.; Parker Jones, O.; Prejawa, S.; Green, D.W.; Price, C.J. Four Functionally Distinct Regions in the Left Supramarginal Gyrus Support Word Processing. Cereb. Cortex 2016, 26, 4212–4226.
- Seghier, M.L. The Angular Gyrus: Multiple Functions and Multiple Subdivisions. Neuroscientist 2013, 19, 43–61.
- Stoeckel, C.; Gough, P.M.; Watkins, K.E.; Devlin, J.T. Supramarginal Gyrus Involvement in Visual Word Recognition. Cortex 2009, 45, 1091–1096.
- Faedo, A.; Tomassy, G.S.; Ruan, Y.; Teichmann, H.; Krauss, S.; Pleasure, S.J.; Tsai, S.Y.; Tsai, M.-J.; Studer, M.; Rubenstein, J.L.R. COUP-TFI Coordinates Cortical Patterning, Neurogenesis, and Laminar Fate and Modulates MAPK/ERK, AKT, and ß-Catenin Signaling. Cereb. Cortex 2008, 18, 2117–2131.
- Barkovich, A.J.; Guerrini, R.; Kuzniecky, R.I.; Jackson, G.D.; Dobyns, W.B. A Developmental and Genetic Classification for Malformations of Cortical Development: Update 2012. Brain 2012, 135, 1348–1369.
- Guerrini, R.; Dobyns, W.B. Malformations of Cortical Development: Clinical Features and Genetic Causes. Lancet Neurol. 2014, 13, 710–726.
- Juric-Sekhar, G.; Hevner, R.F. Malformations of Cerebral Cortex Development: Molecules and Mechanisms. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 293–318.
- Parrini, E.; Conti, V.; Dobyns, W.B.; Guerrini, R. Genetic Basis of Brain Malformations. Mol. Syndromol. 2016, 7, 220–233.
- Hansen, D.V.; Lui, J.H.; Parker, P.R.L.; Kriegstein, A.R. Neurogenic Radial Glia in the Outer Subventricular Zone of Human Neocortex. Nature 2010, 464, 554–561.
- Shitamukai, A.; Konno, D.; Matsuzaki, F. Oblique Radial Glial Divisions in the Developing Mouse Neocortex Induce Self-Renewing Progenitors Outside the Germinal Zone That Resemble Primate Outer Subventricular Zone Progenitors. J. Neurosci. 2011, 31, 3683–3695.
- Vaid, S.; Camp, J.G.; Hersemann, L.; Eugster Oegema, C.; Heninger, A.-K.; Winkler, S.; Brandl, H.; Sarov, M.; Treutlein, B.; Huttner, W.B.; et al. A Novel Population of Hopx-Dependent Basal Radial Glial Cells in the Developing Mouse Neocortex. Development 2018, 145, dev169276.
- Wang, X.; Tsai, J.-W.; LaMonica, B.; Kriegstein, A.R. A New Subtype of Progenitor Cell in the Mouse Embryonic Neocortex. Nat. Neurosci. 2011, 14, 555–561.
- Wong, F.K.; Fei, J.-F.; Mora-Bermúdez, F.; Taverna, E.; Haffner, C.; Fu, J.; Anastassiadis, K.; Stewart, A.F.; Huttner, W.B. Sustained Pax6 Expression Generates Primate-like Basal Radial Glia in Developing Mouse Neocortex. PLoS Biol. 2015, 13, e1002217.
- Fietz, S.A.; Kelava, I.; Vogt, J.; Wilsch-Bräuninger, M.; Stenzel, D.; Fish, J.L.; Corbeil, D.; Riehn, A.; Distler, W.; Nitsch, R.; et al. OSVZ Progenitors of Human and Ferret Neocortex Are Epithelial-like and Expand by Integrin Signaling. Nat. Neurosci. 2010, 13, 690–699.
- Bershteyn, M.; Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Nene, A.; Wynshaw-Boris, A.; Kriegstein, A.R. Human IPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 2017, 20, 435–449.e4.
- Li, Y.; Muffat, J.; Omer, A.; Bosch, I.; Lancaster, M.A.; Sur, M.; Gehrke, L.; Knoblich, J.A.; Jaenisch, R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396.e3.
- Qian, X.; Jacob, F.; Song, M.M.; Nguyen, H.N.; Song, H.; Ming, G. Generation of Human Brain Region–Specific Organoids Using a Miniaturized Spinning Bioreactor. Nat. Protoc. 2018, 13, 565–580.
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De Novo Mutations Revealed by Whole-Exome Sequencing Are Strongly Associated with Autism. Nature 2012, 485, 237–241.
- Autism Sequencing Consortium; Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D’Gama, A.M.; Kim, S.N.; Hill, R.S.; et al. Rates, Distribution and Implications of Postzygotic Mosaic Mutations in Autism Spectrum Disorder. Nat. Neurosci. 2017, 20, 1217–1224.
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative Functional Genomic Analyses Implicate Specific Molecular Pathways and Circuits in Autism. Cell 2013, 155, 1008–1021.
- The DDD Study; Homozygosity Mapping Collaborative for Autism; UK10K Consortium; The Autism Sequencing Consortium; De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Ercument Cicek, A.; et al. Synaptic, Transcriptional and Chromatin Genes Disrupted in Autism. Nature 2014, 515, 209–215.
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A Community-Driven Knowledgebase for the Autism Spectrum Disorders (ASDs). Mol. Autism 2013, 4, 36.
- Cotney, J.; Muhle, R.A.; Sanders, S.J.; Liu, L.; Willsey, A.J.; Niu, W.; Liu, W.; Klei, L.; Lei, J.; Yin, J.; et al. The Autism-Associated Chromatin Modifier CHD8 Regulates Other Autism Risk Genes during Human Neurodevelopment. Nat. Commun. 2015, 6, 6404.
- Watt, N.; Wetherby, A.M.; Barber, A.; Morgan, L. Repetitive and Stereotyped Behaviors in Children with Autism Spectrum Disorders in the Second Year of Life. J. Autism Dev. Disord. 2008, 38, 1518–1533.
- Hobbs, M.M.; Wolters, W.C.; Rayapati, A.O. Bosch-Boonstra-Schaaf Optic Atrophy Syndrome Presenting as New-Onset Psychosis in a 32-Year-Old Man: A Case Report and Literature Review. J. Psychiatr. Pract. 2020, 26, 58–62.
- Molloy, A.; Rowe, F.J. Manneristic Behaviors of Visually Impaired Children. Strabismus 2011, 19, 77–84.
- Kerr, M.; Gil-Nagel, A.; Glynn, M.; Mula, M.; Thompson, R.; Zuberi, S.M. Treatment of Behavioral Problems in Intellectually Disabled Adult Patients with Epilepsy. Epilepsia 2013, 54, 34–40.
- Furniss, F.; Biswas, A.B. Recent Research on Aetiology, Development and Phenomenology of Self-Injurious Behaviour in People with Intellectual Disabilities: A Systematic Review and Implications for Treatment: Aetiology & Phenomenology of SIB in ID: A Systematic Review. J. Intellect. Disabil. Res. 2012, 56, 453–475.
- Inoue, M. Assessments and Interventions to Address Challenging Behavior in Individuals with Intellectual Disability and Autism Spectrum Disorder in Japan: A Consolidated Review. Yonago Acta Med. 2019, 62, 169–181.
- Matson, J.L.; Dempsey, T.; Rivet, T.T. The Interrelationships of Psychopathology Symptoms on Social Skills in Adults with Autism or PDD-NOS and Intellectual Disability. J. Dev. Phys. Disabil. 2009, 21, 39–55.
- Oliver, C.; Licence, L.; Richards, C. Self-Injurious Behaviour in People with Intellectual Disability and Autism Spectrum Disorder. Curr. Opin. Psychiatry 2017, 30, 97–101.
- Smith, K.R.M.; Matson, J.L. Social Skills: Differences among Adults with Intellectual Disabilities, Co-Morbid Autism Spectrum Disorders and Epilepsy. Res. Dev. Disabil. 2010, 31, 1366–1372.
- Lowe, K.; Allen, D.; Jones, E.; Brophy, S.; Moore, K.; James, W. Challenging Behaviours: Prevalence and Topographies. J. Intellect. Disabil. Res. 2007, 51, 625–636.
- Jacob, S.; Landeros-Weisenberger, A.; Leckman, J.F. Autism Spectrum and Obsessive–Compulsive Disorders: OC Behaviors, Phenotypes and Genetics. Autism Res. 2009, 2, 293–311.
- Muehlmann, A.M.; Lewis, M.H. Abnormal Repetitive Behaviours: Shared Phenomenology and Pathophysiology: Abnormal Repetitive Behaviours. J. Intellect. Disabil. Res. 2012, 56, 427–440.
- Tomassy, G.S.; De Leonibus, E.; Jabaudon, D.; Lodato, S.; Alfano, C.; Mele, A.; Macklis, J.D.; Studer, M. Area-Specific Temporal Control of Corticospinal Motor Neuron Differentiation by COUP-TFI. Proc. Natl. Acad. Sci. USA 2010, 107, 3576–3581.
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of Autism. Trends Neurosci. 2008, 31, 137–145.
- Ha, S.; Sohn, I.-J.; Kim, N.; Sim, H.J.; Cheon, K.-A. Characteristics of Brains in Autism Spectrum Disorder: Structure, Function and Connectivity across the Lifespan. Exp. Neurobiol. 2015, 24, 273–284.
- Lee, J.K.; Andrews, D.S.; Ozonoff, S.; Solomon, M.; Rogers, S.; Amaral, D.G.; Nordahl, C.W. Longitudinal Evaluation of Cerebral Growth Across Childhood in Boys and Girls With Autism Spectrum Disorder. Biol. Psychiatry 2020, 90, 286–294.
- Adhya, D.; Swarup, V.; Nagy, R.; Dutan, L.; Shum, C.; Valencia-Alarcón, E.P.; Jozwik, K.M.; Mendez, M.A.; Horder, J.; Loth, E.; et al. Atypical Neurogenesis in Induced Pluripotent Stem Cells From Autistic Individuals. Biol. Psychiatry 2021, 89, 486–496.
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390.
- Port, R.G.; Oberman, L.M.; Roberts, T.P. Revisiting the Excitation/Inhibition Imbalance Hypothesis of ASD through a Clinical Lens. Br. J. Radiol. 2019, 92, 20180944.
- Siegel-Ramsay, J.E.; Romaniuk, L.; Whalley, H.C.; Roberts, N.; Branigan, H.; Stanfield, A.C.; Lawrie, S.M.; Dauvermann, M.R. Glutamate and Functional Connectivity—Support for the Excitatory-Inhibitory Imbalance Hypothesis in Autism Spectrum Disorders. Psychiatry Res. Neuroimaging 2021, 313, 111302.
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression Networks Implicate Human Midfetal Deep Cortical Projection Neurons in the Pathogenesis of Autism. Cell 2013, 155, 997–1007.
- Zhang, K.; Yu, F.; Zhu, J.; Han, S.; Chen, J.; Wu, X.; Chen, Y.; Shen, T.; Liao, J.; Guo, W.; et al. Imbalance of Excitatory/Inhibitory Neuron Differentiation in Neurodevelopmental Disorders with an NR2F1 Point Mutation. Cell Rep. 2020, 31, 107521.
- Paulsen, B.; Velasco, S.; Kedaigle, A.J.; Pigoni, M.; Quadrato, G.; Deo, A.J.; Adiconis, X.; Uzquiano, A.; Sartore, R.; Yang, S.M.; et al. Autism Genes Converge on Asynchronous Development of Shared Neuron Classes. Nature 2022, 602, 268–273.
- Bjørnæs, H.; Stabell, K.; Henriksen, O.; Løyning, Y. The Effects of Refractory Epilepsy on Intellectual Functioning in Children and Adults. A Longitudinal Study. Seizure 2001, 10, 250–259.
- Duncan, J.S. MRI Studies. Do Seizures Damage the Brain? In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2002; Volume 135, pp. 253–261. ISBN 978-0-444-50814-0.
- Mameniškienė, R.; Puteikis, K.; Jasionis, A.; Jatužis, D. A Review of Accelerated Long-Term Forgetting in Epilepsy. Brain Sci. 2020, 10, 945.
- van Rijckevorsel, K. Cognitive Problems Related to Epilepsy Syndromes, Especially Malignant Epilepsies. Seizure 2006, 15, 227–234.
- Shields, W.D. Infantile Spasms: Little Seizures, BIG Consequences. Epilepsy Curr. 2006, 6, 63–69.
- Sutula, T.P.; Hagen, J.; Pitkänen, A. Do Epileptic Seizures Damage the Brain? Curr. Opin. Neurol. 2003, 16, 189–195.
- Pisani, F.; Fusco, C.; Spagnoli, C. Linking Acute Symptomatic Neonatal Seizures, Brain Injury and Outcome in Preterm Infants. Epilepsy Behav. 2020, 112, 107406.
- Sutula, T.P.; Pitkanen, A. More Evidence for Seizure-Induced Neuron Loss: Is Hippocampal Sclerosis Both Cause and Effect of Epilepsy? Neurology 2001, 57, 169–170.
- Scharfman, H.E. The Neurobiology of Epilepsy. Curr. Neurol. Neurosci. Rep. 2007, 7, 348–354.
- Bozzi, Y.; Provenzano, G.; Casarosa, S. Neurobiological Bases of Autism-Epilepsy Comorbidity: A Focus on Excitation/Inhibition Imbalance. Eur. J. Neurosci. 2018, 47, 534–548.
- Powell, E.M.; Campbell, D.B.; Stanwood, G.D.; Davis, C.; Noebels, J.L.; Levitt, P. Genetic Disruption of Cortical Interneuron Development Causes Region- and GABA Cell Type-Specific Deficits, Epilepsy, and Behavioral Dysfunction. J. Neurosci. 2003, 23, 622–631.
- Lévesque, M.; Biagini, G.; Avoli, M. Neurosteroids and Focal Epileptic Disorders. Int. J. Mol. Sci. 2020, 21, 9391.
- Lodato, S.; Tomassy, G.S.; De Leonibus, E.; Uzcategui, Y.G.; Andolfi, G.; Armentano, M.; Touzot, A.; Gaztelu, J.M.; Arlotta, P.; Menendez de la Prida, L.; et al. Loss of COUP-TFI Alters the Balance between Caudal Ganglionic Eminence- and Medial Ganglionic Eminence-Derived Cortical Interneurons and Results in Resistance to Epilepsy. J. Neurosci. 2011, 31, 4650–4662.
- Touzot, A.; Ruiz-Reig, N.; Vitalis, T.; Studer, M. Molecular Control of Two Novel Migratory Paths for CGE-Derived Interneurons in the Developing Mouse Brain. Development 2016, 143, 1753–1765.
- Verhoog, Q.P.; Holtman, L.; Aronica, E.; van Vliet, E.A. Astrocytes as Guardians of Neuronal Excitability: Mechanisms Underlying Epileptogenesis. Front. Neurol. 2020, 11.
- Anwar, H.; Khan, Q.U.; Nadeem, N.; Pervaiz, I.; Ali, M.; Cheema, F.F. Epileptic Seizures. Discov. Craiova Rom. 2020, 8, e110.
- del Pino, I.; Tocco, C.; Magrinelli, E.; Marcantoni, A.; Ferraguto, C.; Tomagra, G.; Bertacchi, M.; Alfano, C.; Leinekugel, X.; Frick, A.; et al. COUP-TFI/Nr2f1 Orchestrates Intrinsic Neuronal Activity during Cortical Area Patterning. bioRxiv 2019.
- Wu, X.; Li, H.; Huang, J.; Xu, M.; Xiao, C.; He, S. Regulation of Axon Initial Segment Diameter by COUP-TFI Fine-Tunes Action Potential Generation. Neurosci. Bull. 2021, 1–14.
- Aghakhani, Y. The Role of Periventricular Nodular Heterotopia in Epileptogenesis. Brain 2005, 128, 641–651.
- Khoo, H.M.; Gotman, J.; Hall, J.A.; Dubeau, F. Treatment of Epilepsy Associated with Periventricular Nodular Heterotopia. Curr. Neurol. Neurosci. Rep. 2020, 20, 1–11.
- Meroni, A.; Galli, C.; Bramerio, M.; Tassi, L.; Colombo, N.; Cossu, M.; Lo Russo, G.; Garbelli, R.; Spreafico, R. Nodular Heterotopia: A Neuropathological Study of 24 Patients Undergoing Surgery for Drug-Resistant Epilepsy. Epilepsia 2009, 50, 116–124.
- Valton, L.; Guye, M.; McGonigal, A.; Marquis, P.; Wendling, F.; Régis, J.; Chauvel, P.; Bartolomei, F. Functional Interactions in Brain Networks Underlying Epileptic Seizures in Bilateral Diffuse Periventricular Heterotopia. Clin. Neurophysiol. 2008, 119, 212–223.
- Michaud, J.L.; Lachance, M.; Hamdan, F.F.; Carmant, L.; Lortie, A.; Diadori, P.; Major, P.; Meijer, I.A.; Lemyre, E.; Cossette, P.; et al. The Genetic Landscape of Infantile Spasms. Hum. Mol. Genet. 2014, 23, 4846–4858.
- Balciuniene, J.; DeChene, E.T.; Akgumus, G.; Romasko, E.J.; Cao, K.; Dubbs, H.A.; Mulchandani, S.; Spinner, N.B.; Conlin, L.K.; Marsh, E.D.; et al. Use of a Dynamic Genetic Testing Approach for Childhood-Onset Epilepsy. JAMA Netw. Open 2019, 2, e192129.
- Dimassi, S.; Labalme, A.; Ville, D.; Calender, A.; Mignot, C.; Boutry-Kryza, N.; de Bellescize, J.; Rivier-Ringenbach, C.; Bourel-Ponchel, E.; Cheillan, D.; et al. Whole-Exome Sequencing Improves the Diagnosis Yield in Sporadic Infantile Spasm Syndrome. Clin. Genet. 2016, 89, 198–204.
- Hino-Fukuyo, N.; Kikuchi, A.; Arai-Ichinoi, N.; Niihori, T.; Sato, R.; Suzuki, T.; Kudo, H.; Sato, Y.; Nakayama, T.; Kakisaka, Y.; et al. Genomic Analysis Identifies Candidate Pathogenic Variants in 9 of 18 Patients with Unexplained West Syndrome. Hum. Genet. 2015, 134, 649–658.
- Hino-Fukuyo, N.; Kikuchi, A.; Yokoyama, H.; Iinuma, K.; Hirose, M.; Haginoya, K.; Niihori, T.; Nakayama, K.; Aoki, Y.; Kure, S. Long-Term Outcome of a 26-Year-Old Woman with West Syndrome and an Nuclear Receptor Subfamily 2 Group F Member 1 Gene (NR2F1) Mutation. Seizure 2017, 50, 144–146.
- Fournier, K.A.; Hass, C.J.; Naik, S.K.; Lodha, N.; Cauraugh, J.H. Motor Coordination in Autism Spectrum Disorders: A Synthesis and Meta-Analysis. J. Autism Dev. Disord. 2010, 40, 1227–1240.
- Jeste, S.S.; Geschwind, D.H. Clinical Trials for Neurodevelopmental Disorders: At a Therapeutic Frontier. Sci. Transl. Med. 2016, 8, 321fs1.
- Wilson, R.B.; Enticott, P.G.; Rinehart, N.J. Motor Development and Delay: Advances in Assessment of Motor Skills in Autism Spectrum Disorders. Curr. Opin. Neurol. 2018, 31, 134–139.
- Gerber, R.J.; Wilks, T.; Erdie-Lalena, C. Developmental Milestones: Motor Development. Pediatr. Rev. 2010, 31, 267–277.
- Mosconi, M.W.; Kay, M.; D’Cruz, A.M.; Seidenfeld, A.; Guter, S.; Stanford, L.D.; Sweeney, J.A. Impaired Inhibitory Control Is Associated with Higher-Order Repetitive Behaviors in Autism Spectrum Disorders. Psychol. Med. 2009, 39, 1559–1566.
- Donkelaar, H.J.; Lammens, M.; Wesseling, P.; Hori, A.; Keyser, A.; Rotteveel, J. Development and Malformations of the Human Pyramidal Tract. J. Neurol. 2004, 251, 1429–1442.
- Roessmann, U.; Hori, A. Agyria (Lissencephaly) with Anomalous Pyramidal Crossing. Case Report and Review of Literature. J. Neurol. Sci. 1985, 69, 357–364.
- Chow, C.W.; Halliday, J.L.; Anderson, R.M.D.; Danks, D.M.; Fortune, D.W. Congenital Absence of Pyramids and Its Significance in Genetic Diseases. Acta Neuropathol. 1985, 65, 313–317.
- Marcorelles, P.; Laquerriere, A. Neuropathology of Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 109–119.
- Welniarz, Q.; Dusart, I.; Roze, E. The Corticospinal Tract: Evolution, Development, and Human Disorders. Dev. Neurobiol. 2017, 810–829.
- Tocco, C.; Øvsthus, M.; Bjaalie, J.G.; Leergaard, T.B.; Studer, M. The Topography of Corticopontine Projections Is Controlled by Postmitotic Expression of the Area-Mapping Gene Nr2f1. Development 2022, 149, dev200026.
- Greig, L.C.; Woodworth, M.B.; Galazo, M.J.; Padmanabhan, H.; Macklis, J.D. Molecular Logic of Neocortical Projection Neuron Specification, Development and Diversity. Nat. Rev. Neurosci. 2013, 14, 755–769.
- Contesse, T.; Ayrault, M.; Mantegazza, M.; Studer, M.; Deschaux, O. Hyperactive and Anxiolytic-like Behaviors Result from Loss of COUP-TFI/Nr2f1 in the Mouse Cortex. Genes Brain Behav. 2019, 18, e12556.
- Iverson, J.M. Developing Language in a Developing Body: The Relationship between Motor Development and Language Development. J. Child Lang. 2010, 37, 229–261.
- Esposito, G.; Venuti, P.; Maestro, S.; Muratori, F. An Exploration of Symmetry in Early Autism Spectrum Disorders: Analysis of Lying. Brain Dev. 2009, 31, 131–138.
- Bhat, A.N.; Landa, R.J.; Galloway, J.C. Current Perspectives on Motor Functioning in Infants, Children, and Adults with Autism Spectrum Disorders. Phys. Ther. 2011, 91, 1116–1129.
- Karasik, L.B.; Tamis-Lemonda, C.S.; Adolph, K.E. Transition from Crawling to Walking and Infants’ Actions with Objects and People. Child Dev. 2011, 82, 1199–1209.

