1.
Optic Atrophy (OA), Optic Nerve Hypoplasia (ONH) and Optic Disc Abnormalities in Bosch–Boonstra–Schaaf Optic Atrophy Syndrome (BBSOAS)) Patients
In light of its several and variable clinical features affecting the visual system, such as OA or optic nerve pallor, ONH or small optic discs, cerebral visual impairment (CVI), nystagmus (uncontrolled eye movements) and alacrima (decreased tear reflex), BBSOAS can also be defined as a congenital optic neuropathy. One of the main and first-reported visual impairments in BBSOAS children is OA, as the name of the syndrome suggests. It can be defined as optic nerve (ON) damage anywhere from the retina to the lateral geniculate nucleus of the thalamus, usually caused by retinal ganglion cell (RGC) death and retraction of RGC axons, with a resulting pale ON in fundoscopy.
However, OA and ONH have both been reported since the first description of BBSOAS patients [1]. As OA, ONH is also characterized by a deficiency of RGCs and their axons, leading to ganglion cell layer disorganization and a small optic disc with a thin ON. A main difference between ONH and OA is that while ONH is a congenital, non-progressive disease characterized by underdevelopment of the ON, OA is instead degenerative with a normal early development of the ON that deteriorates over time. according to experimental data in Nr2f1-deficient mouse models, BBSOAS-like visual impairments, such as retinal and ON anomalies, result from primary RGC patterning, myelination and inflammatory defects originating during early development (Figure 1) [2][3], and leading to visual system defects that would remain stable during postnatal life [3]. Consistently with early appearance of BBSOAS, ONH could be the major clinical feature accounting for the non-progressive visual dysfunction observed in patients [3].
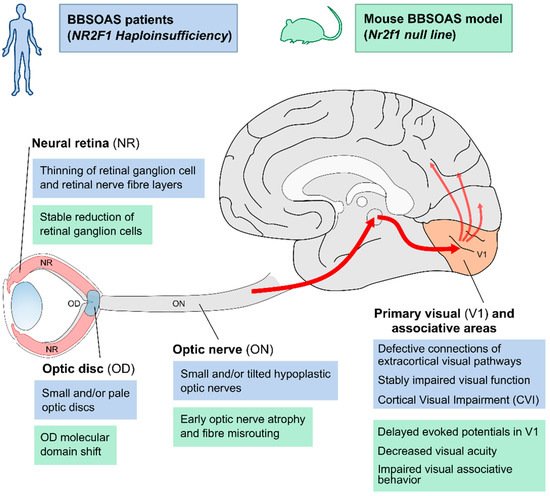 Figure 1. Overview of structural and functional defects along the visual pathway in BBSOAS patients and corresponding Nr2f1 mutant mouse models.
Figure 1. Overview of structural and functional defects along the visual pathway in BBSOAS patients and corresponding Nr2f1 mutant mouse models. CVI and other visual impairments reported in BBSOAS patients (blue boxes) might build upon structural impairment affecting several structures in the visual system, such as the neural retina (NR), the optic disc (OD), the optic nerve (ON), the primary visual area of the neocortex (V1) and its connections to secondary associative areas. The use of
Nr2f1 mutant mouse models (green boxes) have helped in elucidating the molecular, cellular and functional mechanisms that could potentially cause the defects observed in patients.
In addition, several BBSOAS patients show small malformations of the optic disc (OD) at the edge between the neural retina and the ON. Such malformations manifest in excavated, pale or small ODs. Often associated with other congenital eye malformations, OD lesions can negatively affect visual acuity. Interestingly, OD malformations are reported more frequently in patients with NR2F1 deletions (33%) than in patients with other variants (12–22%). A structural change in the form of pale and/or excavated ODs is diagnosed by ophthalmologists as an indirect sign of underlying ON diseases, including OA or ONH. In fact, OD pallor and abnormal shape can result from RGC death (causing axonal fibers loss) and degeneration of pial capillaries entering the optic nerve head In addition, several BBSOAS patients show small malformations of the optic disc (OD) at the edge between the neural retina and the ON. Such malformations manifest in excavated, pale or small ODs. Often associated with other congenital eye malformations, OD lesions can negatively affect visual acuity. Interestingly, OD malformations are reported more frequently in patients with NR2F1 deletions (33%) than in patients with other variants (12–22%). A structural change in the form of pale and/or excavated ODs is diagnosed by ophthalmologists as an indirect sign of underlying ON diseases, including OA or ONH. In fact, OD pallor and abnormal shape can result from RGC death (causing axonal fibers loss) and degeneration of pial capillaries entering the optic nerve head [4][5][6]. However, some OD malformations—such as optic disc coloboma—can be the direct consequence of genetic or environmental insults affecting the development of this region, rather than a consequence of ON disease [7][8][9]. The OD pit, for example, consisting in a round or oval localized depression within the OD, is caused by defective occlusion of the embryonic ventral fissure of the optic vesicle [10][11]. By homology, data from Nr2f1 mouse models showed delayed ventral fissure closure in null embryos [2] or severe coloboma in double Nr2f1 and Nr2f2 conditional mutants [12], suggesting that impaired ventral fissure fusion could be responsible for OD malformations in a Nr2f1-deficient context. This would also fit with reports of BBSOAS patients showing coloboma-like malformations -deficient context. This would also fit with reports of BBSOAS patients showing coloboma-like malformations [13][14][15].
Furthermore, data in mice revealed a molecular network controlled by
Nr2f1 and driving the establishment of the border between the Pax2-expressing optic stalk (for example, the presumptive OD) and the Pax6-expressing retinal vesicle (for example, the presumptive neural retina - NR), ultimately allowing the development of the OD between these two regions
(Figure 13)
[121][16]. One key function of the OD is to produce signaling molecules for RGC axonal guidance, such as Netrin-1
[17][18].
Nr2f1 constitutive mutants demonstrated how the OD genetic network is disrupted by loss of
Nr2f1 alone, resulting in
Pax6 overexpression at the expense of the Pax2 domain
[219]. This results in a shift of the border between NR and OS, with heavy consequences for
Netrin-1 expression, ultimately impacting axonal guidance of RGC fibers exiting the eyeball
[320]. However, defects in
Nr2f1 haploinsufficient animals appear quite subtle compared to OD malformations in BBSOAS patients, suggesting that additional factors could impact OD development in humans and/or that species-specific differences are present.
The involvement of
Pax2 in an
Nr2f1-regulated network is particularly important, as human
PAX2 mutations lead to coloboma-like OD malformations in the context of syndromic conditions
[821][1922][2023], consistent with a key role of the
Nr2f1-Pax2-Pax6 genetic network for the correct establishment of the OD region. Human
NR2F1 could indeed control OD development by modulating
PAX2 and
PAX6, as it does in mouse, but further experiments on human cells are necessary to verify the evolutionary conservation of such a network.
2. When Intellectual Disability Meets Visual Disfunction: Cortical Visual Impairment (CVI) in BBSOAS Patients
The condition of CVI (affecting around 42% of BBSOAS patients, and particularly common in those bearing DBD variants) is a bilateral visual impairment due to a non-ocular cause in the presence of normal pupil reactivity and characterized by abnormal perception, elaboration and interpretation of visual stimuli
[2124][2225]. Despite the presence of nystagmus, ON atrophy and other structural eye anomalies, the degree of visual impairments in BBSOAS patients with CVI exceeds what would be expected from eye abnormalities alone, implying that higher-order visual centers in the brain (such as the retro-chiasmatic visual pathways, the thalamus, the primary cortex and/or the secondary associative visual cortices) might also be affected
[222][2312][24][25][26][27][28][29][30][31]. In BBSOAS patients, poor visual acuity and visual field abnormalities support the diagnosis of CVI. For instance, patients have narrowed visual fields, low visual acuity, and difficulties with distance viewing, following fast moving scenes or recognizing objects in crowded environments
(Table 1). Among the possible factors contributing to CVI in BBSOAS, impaired thalamic connection with parietal and occipital cortices has been hypothesized to impact visuospatial ability
[3132], together with aberrant connections of major fasciculi relaying the occipital and temporal lobes to adjacent associative areas
[320].
Table 1. List of BBSOAS main features. Table resuming the main clinical features of BBSOAS patients with
NR2F1 haploinsufficiency; some of them are often present at birth (congenital), including hypotonia, nystagmus and oromotor dysfunction. The clinical features of BBSOAS are variable, and not every individual necessarily manifests all features. Further, the severity of the condition varies from one individual to the next. Abbreviations: ASD, autism spectrum disorder; ID, intellectual disability; IQ, intelligence quotient; MRI, magnetic resonance imaging.
| BBSOAS Main Feature(s) |
Clinical Description(s) |
| Developmental delay (DD) |
Delay in reaching language, social or motor skills milestones |
| Intellectual disability (ID) |
Significantly reduced ability to understand new or complex information and to learn and apply new skills (impaired intelligence).
IQ ranging from profound ID with IQ < 20, to moderate (35 < IQ < 49) or mild ID (50 < IQ < 69) |
| Visual impairment |
Optic nerve abnormalities and/or brain-based vision impairment: |
| Optic nerve atrophy or pallor |
| Optic nerve hypoplasia |
| Cortical visual impairment (difficulty locating objects in a crowded field and following rapidly moving images and scenes). |
| Alacrima (abnormal amount of reflex tearing) |
| Manifest latent nystagmus and poor tracking; congenital |
| Significant refractive errors |
| Amblyopia |
| Hypotonia |
Low muscle tone; congenital |
| Oromotor dysfunction |
Swallowing, sucking and chewing problems; congenital |
| Repetitive behavior |
Hand flapping, head banging and more |
| Autism spectrum disorder (ASD) |
ASD or autistic traits |
| Seizures |
Infantile and/or febrile; occipital seizures |
| Attention-deficit hyperactivity disorder (ADHD) |
Inattention, impulsivity and hyperactivity |
| Hearing impairment |
Abnormal hearing |
| Dysmorphic facial features |
Mild and inconsistent |
| Thin corpus callosum and neocortical dysgyria |
Hypoplasia of the corpus callosum and abnormal pattern of cortical convolutions and sulci (dysgyria in temporal and parietal areas) on brain MRI |
Nr2f1 mouse mutant models challenged the involvement of central thalamic and neocortical structures at the anatomical, electrophysiological and behavioral levels
(Figure 3). The size of visual thalamic nuclei is affected upon
Nr2f1 loss, in turn acting on the maturation of primary and secondary visual areas as a result of impaired afferent thalamocortical innervation
[32][33][34][35]. At cortical level, the
Nr2f1 high caudolateral to low rostromedial expression gradient is key for a dose-dependent establishment of distinct neocortical areal identities, a process termed arealization
[3536][3637]. Upon mouse
Nr2f1 loss, the rostral motor area expands at the expense of caudal sensory ones, so that the primary visual area (V1) tends to be compressed caudally in
Nr2f1 mutant neocortex, with a striking effect in
null (homozygous) brains and a less pronounced area shift in heterozygous
HET animals
[3233][3334][3738]. Electrophysiological recordings demonstrated a delay in transmitting peripheral visual stimuli along the visual pathway, together with a decrease in amplitude in the visual thalamic nucleus, the superior colliculi and the V1 superficial cortical layers of
HET mice
[219][320]. Suboptimal conduction velocity of visual stimuli along the visual pathway could depend on ONH associated with hypomyelination and gliosis
[219]. Furthermore, visual acuity is decreased in
Nr2f1 HET mice compared to
wild-type littermates, in line with low visual acuity and nondegenerative vision loss in BBSOAS patients
[120][326][1439][2340]. Finally, CVI could also result from impaired elaboration of visual information in secondary associative cortices. Interestingly, while
Nr2f1 haploinsufficient mice are still able to learn and execute complex tasks
[213][3819], they nevertheless fail to associate the visual stimulus with a specific operating task when challenged with a light-dependent operating procedure, somehow recapitulating a deficit in the interpretation of visual stimuli reminiscent of patients’ CVI
[219]. This suggests a specific impairment in the perception and elaboration of visual stimuli in high-order cortices, rather than a generalized defect in learning and cognitive function. Altogether, mouse models point to a scenario in which multiple structures along the visual pathway are impacted by
Nr2f1-deficiency, from the retina and ON up to the thalamus, the V1 and visual associative cortices together with their connecting tracts.
Interestingly, human
NR2F1 shows similar graded expression along neocortical axes
[3214][33][3934][4041], but it is still unknown whether BBSOAS patients display analogous defects in the positioning, size and function of primary and secondary visual areas; functional magnetic resonance imaging (fMRI) analysis and brain tractography could help to tackle this issue in future clinical ones. Undoubtedly, impaired visual learning and aberrant visual processing due to reduced
NR2F1 dosage constitute one of the main features of BBSOAS intellectual deficits, and there is an urgent need of specific early therapeutic interventions to help BBSOAS patients in reaching visual developmental milestones.
3. The Many Converging Roads of Intellectual Disability in BBSOAS Patients: From Corpus-Callosum Thinning to Hippocampal and Neocortical Malformations
The second-most prevalent condition in BBSOAS is a moderate to severe intellectual disability (ID)
[2326], found in 87% of patients and particularly common upon DBD mutations (94% of patients). Interestingly, even the milder BBSOAS genetic categories, such as patients bearing
ligand-binding domain (LBD
) variants or deletions, show high incidence of ID and speech delay (70% and 80% of described patients, respectively). Despite a high frequency of cognitive symptoms, brain morphological malformations underlying ID in BBSOAS patients are still poorly characterized. Due to the high prevalence of visual deficits in BBSOAS, clinicians tend to focus more on optical features when analyzing MRI brain scans
[139][1440]. Moreover, the difficulty in obtaining high-resolution MRI scans from young patients also hampers the acquisition of morphological data. Nevertheless, morphological brain malformations have started to emerge, such as a thin corpus callosum (CC) with general presentation or restricted to posterior regions, hippocampal malrotation or dysmorphia, rare white matter loss (demyelination in 14% patients) and
of localized dysgyria or megalencephaly.
The
corpus callosum (CC
) is the largest commissure connecting brain hemispheres, allowing integration between the two cerebral halves. Notably, CC thinning or agenesis could directly contribute to ID, visual problems, motor impairment, speech delay and seizures described in patients
[41][42][43][44]. Consistently, mouse data have demonstrated the role of
Nr2f1 during CC formation as a regulator of the differentiation and migration of late-born cortical neurons
[4445]. Callosal
Nr2f1-deficient neurons, found in reduced numbers at postnatal ages, also fail to elongate their axons and topographically innervate the contralateral hemisphere
[4445][4546], ultimately leading to a thinning of the CC, as in human patients. Furthermore, upon fiber elongation via axonal growth, long-range tracts, such as the CC, must be properly myelinated for optimal signal conduction; impaired myelination is associated with human clinical conditions, such as demyelinating diseases or white matter disorders leading to ID, seizures, lack of coordination and other neurocognitive consequences
[4647]. In mutant mouse models,
Nr2f1 regulates myelination levels in the ON and CC
[219][4748], similarly to what is observed in BBSOAS patients, suggesting a key role for
Nr2f1 in the maturation of oligodendrocytes and resulting myelination process.
Alongside CC thinning, specific morphological hippocampal defects have been reported in some BBSOAS patients. Since the hippocampus is a key structure for learning, memory and other cognitive processes
[483][49][50] and defects in its development can negatively impact such cognitive performances
[51][52][53][54], it is reasonable to hypothesize a hippocampal component underlying ID in BBSOAS patients. Mouse experiments support this hypothesis, as
Nr2f1 mutants show defective hippocampal morphogenesis and function
[3813][55][56]. By regulating both hippocampal progenitor proliferation and neuronal migration during embryonic and early post-natal development
[56],
Nr2f1 mainly acts on the development of the dorsal-most hippocampal regions
[3813], which are involved in spatial navigation, learning and memory
[57]. In addition, electrophysiological data revealed impairment of two cellular correlates of learning and memory, long-term potentiation and long-term depression in a
Nr2f1 HET mouse model
[55], suggesting that altered synaptic plasticity may also contribute to BBSOAS intellectual impairment. However, mutant mice also manifest a prolonged fear-memory retention
[55], which is in line with unusually good long-term memory reported in up to 75% of BBSOAS patients
[2326]. This suggests that
NR2F1 might play an important role in the retention of memory in both mouse and human hippocampus. The exact impact of hippocampal abnormalities on BBSOAS patients is still unknown to date, and more will be necessary to further assess the contribution of other cortical or subcortical regions.
Recently, a novel cohort of six BBSOAS children showed specific cortical morphological defects, such as polymicrogyria- or dysgyria-like malformations in the supramarginal and angular gyri
[58], regions specifically involved in language, vision, spatial cognition, memory retrieval, attention and number processing
[59][60][61]. Such malformations were sometimes associated with elongated occipital convolutions, reminiscent of a mild caudal megalencephaly
[58]. In mice,
Nr2f1 fine-tunes neural proliferative potential by controlling cell-cycle progression and balancing progenitor pool amplification and local neurogenesis
[58][62]. Upon
Nr2f1 loss, a delay in neurogenesis results in amplification of distinct progenitor classes, which ultimately causes caudal cortical expansion
[58] and increased overall neocortical volume
[55]. The occipital expansion reproduced in mice is somehow reminiscent of the elongated occipital convolutions and megalencephaly reported in some BBSOAS patients, suggesting that common cellular and molecular mechanisms might be shared between mouse and human.
Both the presence of cortical malformations in BBSOAS patients and data coming from mouse models link the
NR2F1 gene to a heterogeneous group of Neurodevelopmental Disorders (NDDs), called malformations of cortical development (MCDs). MCDs comprise a variable class of morphological abnormalities, such as polymicrogyria and macrocephaly, often associated with ID, autism, speech and motor difficulties and/or epilepsy
[4][63][64][65][66]. Hence, these malformations could underlie some of the key BBSOAS cognitive disorders. However, whether they are a common feature of BBSOAS is still unknown, and a more detailed examination of MRI scans in larger cohorts, with a special focus on occipital, parietal and temporal regions, will be needed in future analysis. Furthermore, mouse models could only partially recapitulate these morphological defects
[58], as they lack specific human-like features, such as cortical convolutions and abundance of a specific class of neural progenitor termed basal radial glia, highly present in gyrencephalic species
[66][67][68][69][70][71].
InFor gyrencephalic mammals, such as the ferret
[7271], or in human-like systems such as the induced pluripotent stem cell (iPSC)-derived brain organoids
[72][73][74][75], could help unravel
NR2F1 function in mammalian neural proliferation and neocortex gyrification.
4.
NR2F1
as an Autism Spectrum Disorder Gene
Autism spectrum disorder (ASD) or autistic traits constitute another major clinical feature of BBSOAS
[2326]. ASD is a complex group of NDDs characterized by qualitative impairments in social interaction and communication, with repetitive/stereotyped patterns of behavior, interests and activities. Several genomic ones have unveiled a link between disruptive variants in
NR2F1 gene and high susceptibility to ASD phenotypes
[75][76][77][78][79]. As a result,
NR2F1 is now classified as an ASD gene with “suggestive evidence” in the SFARI (Simons Foundation Autism Research Initiative) database
[75][7679][80][81]. However, while some BBSOAS patients officially meet the requirements for an ASD diagnosis (38% diagnosed with ASD), some of them have at least autistic features (14.1% patients with autistic features but no official ASD diagnosis)
[2326], such as repetitive/stereotyped movements in the form of head-banging and hand-flapping, repetitive language, circumscribed interests and self-injurious behaviors, among others
[8281]. The highly variable behavioral phenotype of BBSOAS also includes attention deficit hyperactivity disorder (ADHD), obsessive compulsive disorder (OCD), pervasive developmental disorder-not otherwise specified (PDD-NOS) and, less commonly, psychosis in the form of auditory hallucinations
[8382].
Due to the limited number of reported patients, together with the variable expressivity of the BBSOAS behavioral spectrum, it is challenging to distinguish behavioral abnormalities due to ASD or to other clinical features, such as visual impairment
[8483], epilepsy
[8584] or ID
[865][876][887][8985]. As an example, children with visual impairments might display head-banging behaviors
[8483]. Furthermore, some stereotyped movements in patients with ID and concurrent epilepsy could be the result of unrecognized seizure activity
[8584], and in general, higher rates of disruptive and self-injurious behavior can be found in patients with combined ASD and epilepsy
[908]. Additionally, ID is often associated with stereotyped and aggressive behaviors
[908][919]. Finally, obsessive-compulsive behaviors typical of ASD and present in some BBSOAS patients partially overlap with typical symptoms associated with OCD
[9210]. The most probable scenario for BBSOAS is that multiple cognitive impairments—ASD, ID and epilepsy, among others—could converge on a common (although variable) behavioral spectrum, and overlapping pathologies could synergistically lead to shared phenotypes
[9311]. Hence, to better discern the different aspects of cognitive impairments characteristic of the BBSOAS behavioral spectrum, further neuropsychological assessments are needed.
As for other BBSOAS features, the use of mouse models has helped to unravel possible etiological mechanisms underlying patients’ autistic features. For instance, some cortical morphological changes reported in
Nr2f1 mutant mice are the result of a regional-specific control of neurogenesis, which, when impaired, might lead to local megalencephaly and/or abnormal gyrification
[58][62]. Moreover, morphological changes are also associated with altered neocortical identity upon
Nr2f1 loss, with posterior sensory areas being reduced in size, and a large cortical surface acquiring anterior motor-like properties
[3315][9434]. Interestingly, arealization defects—especially concerning the frontal lobe—regionally altered gyrification of cortical areas and imbalanced neurogenesis leading to early expansion of the brain have all been linked with ASD in patients
[9586][9687][9788]. Hence, BBSOAS autistic features and other cognitive impairments could be influenced by impaired arealization upon human
NR2F1 perturbations, but further investigation by fMRI is required to investigate potential areal functional impairments.
Another interesting hypothesis is that ASD and other neurodevelopmental impairments described in BBSOAS patients could derive from an excitatory/inhibitory (E/I) dysregulation due to an imbalance between dorsally generated excitatory glutamatergic neurons and ventrally generated inhibitory interneurons
[9889][9990][10091][10192][10293]. In this respect, a recent report introduced a patient-specific DBD variant in mouse
Nr2f1, in an attempt to reproduce a genuine point-mutation mouse model of BBSOAS
[10394]. Mutated
Nr2f1 promoted differentiation of inhibitory neurons, while concomitantly reducing the rate of production of glutamatergic ones, resulting in autistic-like behavioral deficits, such as impaired social interaction. Interestingly, the observed behavioral deficits could be partially alleviated by antagonizing the excessive inhibitory synaptic transmission
[10394]. Consistently, mutations affecting other ASD risk genes have been shown to trigger a similar E/I imbalance through distinct molecular pathways ultimately converging into shared neurodevelopmental abnormalities
[10495]. In summary, while the exact molecular and cellular mechanisms leading to autistic phenotypes are still under investigation, it was expected
that NR2F1 to be soon considered a
bona fide ASD gene.
5.
NR2F1
as a Susceptibility Gene for Infantile Epileptic Disorders
Although not always present (46% of BBSOAS patients), another noteworthy BBSOAS feature is the insurgence of various epileptiform pathologies, comprising infantile and febrile spasms, West syndrome and other forms of epilepsy in children
[126][1439][2340]. Particularly common in DBD patients (53%), epileptic features are more rarely reported in patients with LBD variants or deletions (29% and 27%, respectively), suggesting that epilepsy could represent a BBSOAS feature specifically associated with more severe genetic variants. In some situations, altered electroencephalogram (EEG) patterns—often restricted to the occipital hemispheres—were recorded even in the absence of epileptic seizures
[1596]. The presence of early-onset epileptic forms in infancy (such as infantile spasms) is of particular importance, as these so-called “catastrophic epilepsies” are associated with poor neurodevelopmental outcome and could inflict additional damage to the developing brain, contributing to ID, memory impairment, attention deficits, developmental delay and autistic features
[10597][10698][10799][108100][109101][110102]. The discrimination between brain injuries leading to or caused by seizures has always constituted a great challenge for clinicians
[111103][112104].
The disruption of distinct developmental processes has been hypothesized to underlie the onset of epilepsy
[113105]. As an example, an imbalance of excitatory pyramidal neurons and inhibitory interneuron subtypes has been proposed as common potential cause of epilepsy and autism
[114106][115107]. Alternatively, interneurons could promote seizures in the initial phase of epileptic activity
[116108], suggesting that epileptiform events could originate from GABAergic dysfunction
per se, regardless of their excitatory glutamatergic counterpart. Interestingly, mouse
Nr2f1 is known to regulate the number and type of GABAergic interneurons produced in the ventral telencephalon and reaching the cortex
[117109][118110]. Further, epileptic discharges can also be caused by improper electrophysiological activity of neural circuits due to intrinsic electrical properties of glutamatergic neurons and astrocytes
[119111][120112]. Recently
, it was showed that mouse
Nr2f1 controls the intrinsic electric properties of pyramidal neurons by directly regulating the expression of distinct voltage-gated ion channels as well as the axon initial segment length and diameter
[121113][122114]. By allowing a fine-tuning of action potentials and proper modulation of spontaneous network activity during circuit maturation, mouse
Nr2f1 can ultimately sculpt the emergence of electrical activity in cortical networks
[121113].
Finally, drug-resistant forms of epilepsy can also be caused by nodular periventricular heterotopia
[123115][124116][125117][126118], ectopic clusters of grey matter caused by impairment of neuronal migration and connectivity. Defective migration of neuronal cells has been linked to
Nr2f1 loss in mouse
[4445], possibly due to altered expression of specific cytoskeletal proteins
[4546]. Consistently, periventricular heterotopia has been reported in some BBSOAS patients alongside seizures
[1358][58119], suggesting a cause–effect link between these two phenomena, whereby clusters of misplaced neurons could act as triggering foci.
The
NR2F1 gene is starting to emerge from unbiased exome-sequencing approaches in individuals with epilepsy
[127120][128121][129122][130123], and is associated with West syndrome
[131124], a severe form of infancy epilepsy characterized by clusters of spasms. It was proposed that the inclusion of
NR2F1 in the diagnostic NGS gene panels for epilepsy, which could help families in their diagnostic odyssey to efficient clinical assignment.
6.
NR2F1
on the Move: Motor Dysfunction in BBSOAS Patients
The development of the motor system is critical for an individual to experience the environment and engage in social interactions. In NDDs, motor skill impairments are particularly prevalent, to the extent that they are considered by clinicians as one of the first signs of atypical development
[132125][133126][134127]. BBSOAS patients have frequently been reported as presenting several motor defects, including delayed motor development, stereotyped and repetitive movements and reduced sensorimotor precision and speed, also resulting in defective execution of fine skilled motor behaviors
[2326][3132]. In children, the typical milestones of motor development are reached at 7 months (sitting), at 10 months (crawling) and around 13 months (walking)
[135128]. BBSOAS children instead achieve these milestones much later, at an average age of 14, 16 and 33 months, respectively
[2326]. Interestingly, the incidence of motor delay is higher in patients with DBD variants than those with mutations in other parts of the protein (53% of patients vs. 0% of children bearing all other variants)
[2326]. Notably, the high severity and penetrance of motor impairments in DBD-variant patients could also be the indirect consequence of other concurring clinical features, such as hypotonia and brain damage following epileptic seizures. Moreover, stereotyped and repetitive movements, such as head-banging and self-injurious behaviors, have been reported in several BBSOAS patients
[126][1439][2340]. The etiological origin of stereotypies is still uncertain, but given the high prevalence of ASD among BBSOAS patients and its association with repetitive behaviors
[8281], it is possible that these behaviors represent typical ASD features, rather than separate motor symptoms. Another possibility is that the fronto-striatal circuit, responsible for the inhibition of stereotyped repetitive movements, might also be affected in BBSOAS patients, similarly to what is reported for some ASD patients
[136129].
Abnormal motor behavior can also be observed in the specific context of visual function and eye reflexes. Many BBSOAS patients show nystagmus (repetitive and uncontrolled involuntary movements of the eyeballs), poor object tracking, and saccadic eye movements (rapid, uncontrolled movements of the eyes that abruptly change the point of fixation)
[1426][2340]. In a first thorough characterization of oculomotor skills of a BBSOAS patient, it was showed reduced accuracy when performing specific visual oculomotor tests, compared to age-matched control or ASD individuals
[3132]. Additionally, Bojanek and colleagues performed a series of tests on general manual skills; the BBSOAS patient showed impaired stopping accuracy and reduced reaction time when compared with both ASD or control age-matched individuals, consistent with a global sensorimotor impairment
[3132]. This might suggest weakened modulation of the cerebellum on pontine-brainstem burst cells, whereas the slower reaction times could implicate a dysfunction of either the descending cortico-ponto-cerebellar and/or the ascending cerebellar-thalamo-frontal circuits
[3132]. However, to define the prevalence of these motor dysfunctions among BBSOAS patients and the possible neuroanatomical correlates, a systematic assessment of motor features coupled with MRI examination on bigger cohorts is needed.
In humans, abnormalities of corticopontine and corticospinal descending tracts are usually associated with more broad brain malformations, impacting the execution of voluntary movements and hand dexterity
[137130][138131][139132][140133][141134]. Previous ones in mice helped in describing the development of cortical descending tracts, and noted the existence of several critical steps, each controlled by specific assets of molecular players (revised in
[141134]). The use of different mouse
Nr2f1 mutants suggested altered wiring in the descending corticopontine and corticospinal tracts, and pointed particularly to the topographic organization of corticopontine projections, an essential process for the control of fine voluntary movements in rodents and humans
[142135]. Furthermore, the impairment observed in both tangential (areal) and radial (laminar) organization of the neocortical layers upon cortical specific loss of
Nr2f1 function ultimately results in abnormal connectivity between the cortex and its subcerebral targets
[3315][3734][9438][143136]. This defective connectivity has a deleterious effect on the accuracy of voluntary motor execution and object reaching behavior: although retaining general normal motor capabilities, fine-skilled paw movements show a remarkable impairment in
Nr2f1 mutant animals
[9415], somehow recapitulating fine motor impairments of BBSOAS patients. Finally, it is reasonable to hypothesize that reduced dexterity could be also impacted by hyperactive features, which have been reported in patients
[1426][2340] and described in
Nr2f1 mouse mutants
[144137].
Further clinical reports will be necessary to fully characterize motor dysfunctions in BBSOAS patients and to distinguish abnormalities due to ASD from those primarily dependent on other motor deficits. However, independently of their clinical origin, motor dysfunctions might represent a first sign of atypical development in BBSOAS. In fact, early-onset motor impairments often emerge before social and communicative deficits
[134127][145138][146139][147140]. Hence, they may serve as an early clinical indicator for BBSOAS, as happens for ASD patients
[134127][145138][148141].