 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Justyna Strycharz | + 6508 word(s) | 6508 | 2020-09-21 06:09:49 | | | |
| 2 | Rita Xu | -2759 word(s) | 3749 | 2020-09-28 08:37:30 | | | | |
| 3 | Rita Xu | Meta information modification | 3749 | 2020-09-28 10:00:24 | | | | |
| 4 | Rita Xu | + 523 word(s) | 4272 | 2020-09-29 06:03:30 | | | | |
| 5 | Rita Xu | -517 word(s) | 3755 | 2020-09-29 10:32:03 | | |
Video Upload Options
Metabolic syndrome (MetS) constitutes a cluster of at least three out of five of the conditions including central obesity, high blood pressure, high blood sugar, high serum triglycerides, and low serum high-density lipoprotein (HDL). Patients diagnosed with MetS exhibit hallmarks of redox imbalance while oxidative stress is now perceived as both the cause and the consequence of MetS.
1. Introduction
Metabolic syndrome (MetS) constitutes a cluster of at least three out of five of the conditions including central obesity, high blood pressure, high blood sugar, high serum triglycerides, and low serum high-density lipoprotein (HDL). It is estimated that at least one third of European and American populations and 27% of Chinese population suffer from MetS [1][2]. The most popular definition used for surveys and health care plan is by IDF (International Diabetes Federation) 2006 [3][4]:
Waist > 94 cm (men) or > 80 cm (women) in Europe, > 102 cm (men) or > 88cm (women) in USA, > 90 cm (men) or > 80 cm (women) in Asia, along with the presence of 2 or more of the following:
Blood glucose greater than 5.6 mmol/L (100 mg/dl) or diagnosed diabetes
HDL cholesterol < 1.0 mmol/L (40 mg/dl) in men, < 1.3 mmol/L (50 mg/dl) in women or drug treatment for low HDL-C
Blood triglycerides (TG) > 1.7 mmol/L (150 mg/dl) or drug treatment for elevated triglycerides
Blood pressure > 130/85 mmHg or drug treatment for hypertension (HT)
The incidence of MetS is rising worldwide contributing not only to the increased morbidity and mortality but also to the dramatic increase of treatment costs. Except for pharmacological management, current therapeutic methods include primarily lifestyle changes and diet. Unfortunately, most patients do not follow these recommendations. Bariatric surgery, reserved for patients with morbid obesity, made significant progress in the treatment of obesity and related metabolic disorders, however, it is associated with serious risk and side effects. OxS is a vital phenomenon occurring in plethora of metabolic disorders including type 2 diabetes (T2DM), obesity and cancer [5].
2. Overview of Oxidative Stress
OxS, as a prolonged state of a disbalance between the oxidative and antioxidative systems of the cells, results in the overproduction of free radicals and reactive oxygen species (ROS), e.g., superoxide anion (O2−•), hydroxyl radical (•OH), singlet oxygen (1O2), hydrogen peroxide (H2O2), hypochlorous radical (ClO−), peroxinitrite radical (ONOO−), and nitric oxide (NO)). Nevertheless, O2−• constitutes the precursor for H2O2, •OH, and ONOO− [6]. ROS could attack virtually all types of biological molecules, leading to further cellular and tissue damage. Lipid peroxidation (LPO) is an autocatalytic process generating reactive aldehydes, e.g., malondialdehyde (MDA), trans-4-oxo-2-nonenal (4-ONE) and trans-4-hydroxy-2-nonenal (4-HNE). Protein carbonylation appears due to protein oxidation via ROS (“direct protein carbonylation” of prolines, lysines, threonines, and arginines). The “secondary protein carbonylation” occurs non-oxidatively within side chains of arginines, lysines, and cysteines with adduction of reactive carbonyl species originating from products of LPO as well as autooxidation of carbohydrates (methylglyoxal, glyoxal) [7]. Genotoxic stress is elicited upon oxidative damage to nitrogenous bases within DNA. The most well-known one, 8-oxo-7,8-dihydroguanine (8-oxoG), is perceived as an indicator of whole-body marker of OxS in urine [8].
In general, elevated level of ROS is capable of posing tremendous threat to each cell due to altering its function, metabolism, cell cycle as well as introducing genetic mutations and triggering apoptosis. Importantly, ROS modulate function of cells through modifying proteins at the posttranslational level via phosphorylation and sulfonylation, nitrosylation, carbonylation, and glutathionylation [9][10]. OxS is generated by incomplete reduction of oxygen as electrons flow from one complex to the next in various dynamic intracellular organelles such as the endoplasmic reticulum (ER), lysosomes and mitochondria as by-products of oxidative protein folding, dysfunctional autophagy, mitochondrial respiration and detoxification [5][11]. The essential sources of ROS are the following enzymes: the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase family of enzymes (NOX), as well as cyclooxygenases, xanthine oxidase (XO), myeloperoxidase (MPO), lipooxygenases and nitric oxide synthase (NOS) [12]. Enzymes critical for the strategy of preventing ROS formation are: superoxide dismutases (SODs), glutathione peroxidases (GPx), catalase (CAT) and glutathione reductase (GR) [13].
2.1. The Interplay between Oxidative Stress and Metabolic Syndrome
While it is broadly accepted that OxS has a role in the pathogenesis of MetS, it is the matter of debate about its causal impact [14]. From the less controversial point of view, OxS constitutes both the consequence and the trigger for MetS, forming a pathogenic vicious cycle initiated in hypertrophic WAT [14][15][16]. Patients diagnosed with MetS exhibit serum hallmarks of redox imbalance in the form of, e.g., increased levels of protein oxidation products, MDA, elevated XO activity, hyperglycemia (HG), elevated TG as well reduced concentrations of HDL-C, vitamin E and C along with declined levels of heat shock response proteins (HSP70) and SOD as compared to healthy probands [17][18][19]. Other studies indicated raised activity of erythrocyte-specific SOD and MPO with elevation of plasma concentrations of H2O2 and MDA in MetS patients in comparison to controls [20][21]. Moreover, a cross-sectional study conducted on the limited number of Japanese MetS patients and healthy subjects indicated an increase of systemic OxS, as determined by urinary 8-epiprostaglandin F2α (8-epi-PGF2α) in single urine samples, being correlated with visceral AT (VAT) accumulation [22]. MetS patients, including those with T2DM, also exhibited elevation of plasma thiobarbituric acid reactive substances (TBARS), protein carbonylation products, and NOx, where the latter indicated the phenomenon of nitrosative stress (NS) [23][24][25]. Additionally, study on MetS and healthy subjects reported raised values for advanced oxidation protein products (AOPP) and pro-oxidant-antioxidant balance (PAB), in plasma and serum, respectively [26]. Importantly, regression analysis indicated positive and independent association between MetS and higher PAB values [56]. Another report showed that levels of ischemia modified albumin (IMA), a protein oxidation marker typical of hypoxia and acidosis, and AOPP were increased with the number of risk factors for MetS, yet it was more significant for AOPP. The latter was also revealed as an independent determinant for occurrence of MetS in studied population of Poles [27]. Interestingly, according to data obtained by Venturini et al., AOPP are more related to components of MetS than markers of LPO [28]. The elevated release of O2−• from the monocytes of MetS patients, plasma levels of ox-LDL, and nitrotyrosine as compared with healthy probands was also found [29]. Appealingly, Yubero-Serrano et al. investigated the relationship between OxS degree and the number of components of MetS in patients. They indicated that activity of SOD and GPx was substantially declined in patients suffering from 2 MetS components than probands with 4/5 MetS components [30]. The general relationship between OxS and MetS is presented in Figure 1.
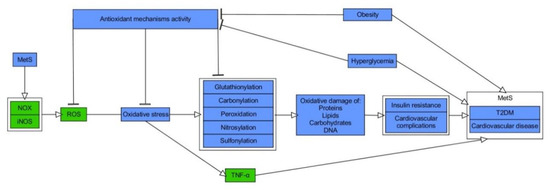
Figure 1. Molecular relationship between oxidative stress and metabolic syndrome. Stimulatory interactions are indicated by arrows and inhibition by T-bars. Actions related to boxes refer to all items inside the box. The figure is a part of paper by Włodarski et al. [31]. MetS–metabolic syndrome, NOX–NADPH oxidase, iNOS–inducible nitric oxide synthase, ROS–reactive oxygen species, TNF-α–tumour necrosis factor-α, T2DM–type 2 diabetes.
2.2. Hypertrophic, Hypoxic and Inflamed White Adipose Tissue—The Initial Fire for Pathogenic Vicious Cycle of Oxidative Stress in Metabolic Syndrome
The pathogenic mechanisms of MetS are complex, thus remaining to be fully elucidated. WAT is a structure comprised mainly of adipose-derived stem cells (ASCs), preadipocytes, adipocytes and immune cells, which is responsible for fat storage. It deposits an excess of energy in triglycerides or mobilizes fatty acids (FA) according to current metabolic needs. It is perceived as an immunological organ and it releases polypeptides (adipo-/cytokines) as well as metabolites capable of exerting systemic actions, including body weight/energy balance, appetite regulation, glucose homeostasis, insulin signaling, and blood pressure control [32][33]. The first known adipocyte hormone, leptin, whose genetic absence causes massive obesity, suppresses appetite, while other hormones, like adiponectin, have just the opposite effect [34][35]. Adiponectin increases sensitivity of cells to insulin as well as pancreatic β-cells survival and functionality. Furthermore, it exerts cardio- and vasculoprotective impact along with regulating the function of macrophages [36]. Interestingly, Benrick A et al. determined that overexpression of adiponectin has a positive influence on WAT. It decreases the size of adipocytes, increases mitochondrial density, and mediates transcriptional upregulation of factors related to efficient esterification of free fatty acids (FFA) [37]. In overall, due to its insulin-sensitizing, antioxidative, anti-inflammatory, and anti-atherogenic impact, adiponectin protects against the MetS [38].
However, the primary trigger for most of the pathways investigated in MetS is adiposity, especially visceral one, thus stressing the importance of a high caloric intake as a major causative factor [39]. Indeed, prolonged and excessive intake of calories, which exceeds white adipocytes’ storage capacity, induces their hypertrophy and hyperplasia, leading to WAT’s hypoxia, along with consequent necrosis and apoptosis of fat cells. These events elicit burst of OxS and M1 type macrophages’ infiltration [40]. Both macrophages and adipocytes produce and secrete proinflammatory adipo-/cytokines and chemokines (e.g., resistin, visfatin, tumour necrosis factor α (TNF-α), monocyte chemoattractant protein-1 (MCP-1, also known as CCL2), interleukin 1β (IL-1β), plasminogen activator inhibitor-1 (PAI-1), interleukin 6 (IL-6), retinol-binding protein 4 (RBP4), and C-reactive protein (CRP)). Hypertrophic WAT is characterized by upregulation of CC chemokines (CCL1/2/3/5,7,8) and their respective receptors (CCR1/2/3/5), the molecules responsible for trafficking leukocytes for the site of inflammation, while CCL2/CCR2 axis is a major one for recruitment of macrophages into WAT [41][42]. Proinflammatory cytokines induce signaling pathways of c-Jun N-terminal kinase (JNK) and IκB kinase-β (IKK-β), while the latter activates a transcription factor involved in production of cytokines and proinflammatory factors, called nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [43][44]. NF-κB is an inducible transcription factor and master regulator of inflammation due to inducing macrophages polarization as well as expression of numerous cytokines (e.g., IL-1, IL-6, TNF-α) and chemokines (e.g., MCP-1, IL-18) [45]. Once secreted, these cytokines activate their extracellular receptors. Simultaneously, the levels of anti-inflammatory adipokines, e.g., adiponectin, omentin decrease. The above biological events trigger a redox imbalance between ROS production and their scavenging, leading to induction of low grade chronic inflammation and intensification of OxS–paving the way for MetS [39][43][46][47]. These phenomena vastly affect WAT, which becomes insulin-resistant, and initiates hyperinsulinemia, enhances lipolysis, increases levels of circulating FFA and their deposition in muscles, liver and pancreas, followed by lipotoxicity, elevated production of glucose due to increased gluconeogenesis and glycogenolysis, and finally, systemic IR and HG [39][48]. Furthermore, atherogenic dyslipidemia, endothelial dysfunction, introduction of hypercoagulable state and HT are observed, thus probably presenting almost the entire spectrum of MetS components [3].
Moreover, the sources of adipose ROS are diversified, being under control of both hormonal and metabolic determinants [49]. Aside from inflammatory cells, mitochondria, as mini factories for ATP production due to oxidative phosphorylation, constitute a major producer of superoxide anions which are converted into H2O2 via SOD2 [50]. It was reported that mitochondrial dysfunction and consequent increased levels of ROS repress insulin signaling as well as production of adiponectin promoting IR in fat cells [51]. Moreover, WAT is the source of ROS-generating enzymes such as NOX, xanthine dehydrogenase/oxidoreductase system (XOR), endoplasmic reticular oxidoreductin 1 (ERO1), pyruvate dehydrogenase (PDH), nicotinamide nucleotide transhydrogenase (NNT) [49]. Interestingly, diet-induced obesity (DIO) in mice supported the elevation of mitochondrial ROS generated by fat cells, thus accelerating mitochondrial uncoupling, biogenesis, and fatty acid oxidation so as to prevent from weight gain and serving as an adaptive mechanism. In other words, the deficiency of SOD2 in adipocytes resulted in increased superoxide levels but simultaneously the lack of IR and increased body mass in spite of obesogenic conditions [52]. Currently, ROS are perceived as second messengers which may facilitate resistance to stress in WAT. However, biological outcomes of short-term and long term-ROS are fully different. For instance, while the first one can be produced upon insulin stimulation, the latter deteriorates insulin response leading to WAT dysfunction [50].
2.3. Insulin Resistance, Hyperglycemia and Oxidative Stress
IR is an impaired response of the body to insulin, resulting in elevated levels of glucose in the blood (a key component of T2DM and MetS) [53]. OxS has been recognized as especially important mechanism in IR [54]. The hormone insulin features a pivotal role in maintaining physiological levels of blood glucose through various effects on insulin target cells [55]. For instance, it elicits vasodilatory as well as vasoconstrictive effects due to the stimulation of endothelial cells for the release of endothelin and nitric oxide, thus increasing the distribution of glucose from blood to organs [56]. Insulin is also critical for highly insulin-sensitive cells, such as muscle, hepatic, and fat ones. Transduction of insulin signal takes place via transmembrane insulin receptors (INSR), whose activation involves dimerization and autophosphorylation of tyrosines located on the intracellular part of receptors due to their kinase activity [55]. Phosphorylated tyrosines are used by adaptor proteins, such as widely known insulin receptor substrates (IRS), as docking sites. These molecules also undergo phosphorylation and mediate the signal via two major pathways: phosphatidyl inositol 3-kinase (PI3K) / protein kinase B (AKT), which activation results in plethora of metabolism-oriented actions, and mitogen-activated kinases (MAPK), which are mainly responsible for growth and differentiation of cells [55]. PI3K conducts phosphatidyl inositol 4,5-biphosphate (PIP2) to phosphatidyl inositol 3,4,5-triphosphate (PIP3) conversion. This is indispensable for plasma membrane recruitment of AKT, followed by phosphorylation of its two specific serine sites by 3-phosphoinositide-dependent kinase-1 (PDK) and mammalian target of rapamycin complex 2 (mTORC 2) [55]. Activated AKT is capable of phosphorylating numerous downstream proteins so as to exert metabolic functions of insulin such as induction of glycogenesis (glycogen synthase (GS)), repression of gluconeogenesis (forkhead box O1 (FOXO1)) or lipolysis (phosphodieterase-3B (PDE-3B)) [44]. Moreover, it initiates intracellular glucose transport due to phosphorylation of Akt substrate of 160 kDa (A160), which is responsible for translocation of glucose transporter 4 (GLUT-4) to cellular membrane in fat and muscle cells [57][58]. IR always involves disturbances in intracellular insulin signaling [59]. It commonly concerns decreased activity or expression of molecules involved in signal transduction (INSR, IRS-1, GLUT-4), decreased expression / translocation of GLUT-4, or increased expression/activity of antagonists of PI3K/AKT pathway, e.g., phosphatase and tensin homolog (PTEN), and polypyrimidine tract binding protein-1 (PTP1B) [60][61][62][63]. The impaired signaling of insulin demands increased concentrations of insulin (hyperinsulinemia). However, an extensive work of pancreatic β-cells to secrete insulin into bloodstream is ultimately pointless, as the vicious cycle of IR is starting to develop, finally leading to a further decrease of available insulin-stimulated GLUT-4 in the cellular membrane and hyperglycemia [64]. Connections between OxS and insulin signaling are illustrated in Figure 2.
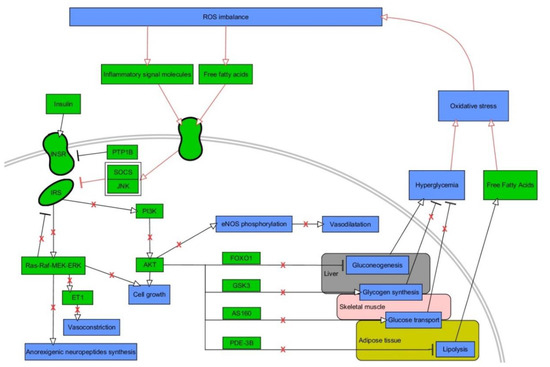
Figure 2. A vicious cycle between oxidative stress and insulin signaling. Stimulatory interactions are indicated by arrows and inhibition by T-bars. Actions related to boxes refer to all items inside the box. Pathological interactions are highlighted in red as well as red crosses denoting withdrawal of physiological insulin signaling. The figure is a part of paper by Włodarski et al. [31]. Processes/phenomena are highlighted in blue and proteins/compounds are highlighted in green. INSR—insulin receptor, IRS—insulin receptor substrate, PTP1B—protein tyrosine phosphatase 1B, SOCS—suppressor of cytokine signaling, JNK–c-Jun N-terminal kinase, PI3K—phosphoinositide 3-kinase, AKT—protein kinase B, FOXO1—forkhead box protein O1, GSK3—glycogen synthase kinase 3, AS160—Akt substrate of 160 kDa, PDE-3B—phosphodiesterase 3 B, eNOS—endothelial nitric oxide synthase.
Lipotoxicity, which is associated with increased plasma level of FFA and intracellular lipid efflux, is vastly involved in muscle and hepatic IR and dysfunction of β-cells [65]. Moreover, FFA stimulate signaling via protein kinase C (PKC) so as to induce NADPH oxidase-mediated OxS and inflammatory signaling via IKK-β and JNK pathways, which perform direct phosphorylation of IRS [66]. The most well-known indicators of lipotoxicity, intracellular lipid intermediates such as diacylglicerol (DAG) and ceramides, act via either different forms of PKC or via protein phosphatase 2A (PPA2) so as to sequester AKT2 or directly affect IRS proteins [65][67]. Accumulating amount of data suggests that ROS impair insulin synthesis and secretion and induce IR [68]. Furthermore, in the course of IR, hyperinsulinemia makes PI3K phosphorylate Rac Family Small GTPase 1 (Rac1) protein instead of PIP2, thus potentiating activity of NOX4 and elevating ROS production [69]. OxS impairs intracellular signaling of insulin due to potentiating activity of SH2-containing tyrosine-protein phosphatase (SHO2), PTP1B and glycogen synthase kinase-3 (GSK-3β) [70][71]. One of the major products of LPO, 4-HNE, potentiates activity of GSK-3β, affects IRS, decreases secretion of adiponectin, and induces lipolysis, protein carbonylation and, finally, IR in muscles [50][72]. Reactive aldehydes make adducts with numerous cellular proteins in various cellular compartments. They mediate protein carbonylation which results in either accumulation or accelerated degradation of affected proteins, enzymes inactivation, changes in gene expression and mitochondrial dysfunction [7]. For instance, several proteins associated with insulin signaling, lipotoxicity, and response to cellular stresses were reported to be carbonylated in WAT of obese insulin-resistant mice, while fatty acid-binding protein was proved to be carbonylated by 4-HNE in vivo [73]. Indeed, protein carbonylation is becoming a more and more studied component of IR and T2DM pathogenesis [7]. Finally, it is worth highlighting that IR is connected with the coordinated interaction among oxidative, nitrosative, genotoxic, carbonyl, and ER stress [53].
HG is a trigger for generation of ROS at the amount which could not be managed by antioxidative system [71]. The central role for HG-mediated OxS is thought to be dependent on the inhibition of glyceraldehyde-3 phosphate dehydrogenase (GAPDH) and accumulation of GAP that upregulates some pathways branching of glycolysis. Namely, HG enhances production of ROS via increasing flux into the polyol, hexosamine and the glyceraldehyde autoxidation pathways as well as activating DAG/PKC signaling pathway, and stimulating formation of advanced glycation end (AGE) products [50][71]. Thus, HG is capable of activating numerous pathways associated with inflammation and OxS, e.g., an activation of PKC stimulates NOX enzymes and lipoxygenases [74]. Excessively generated sorbitol (polyol pathway) mediates activation of p38 MAPK and JNK – core proteins in inflammatory response [75]. The formation of glyoxal (product of glucose autooxidation) and methylglyoxal (product of GAP dephosphorylation) take part in the formation of AGE products. Both precursors bind to specific AGE receptors (RAGE) or interact with various biomolecules thus accelerating OxS via PKC-dependent or independent pathways [71]. Interestingly, methylglyoxal itself affects interaction of insulin with its receptor [76]. AGE/RAGE pathway promotes vascular endothelium’s expression of MCP-1, known to indicate vascular endothelial dysfunction and prothrombotic impact [77]. Furthermore, it is also involved in promoting expression of NF-κB via toll-like receptor 4 (TLR4) pathway [54]. It is to be noted that the early glycation of proteins can be also reversible, as in Schiff bases or Amadori adducts, including a marker of diabetes, glycated hemoglobin (HbA1C) [78]. For thoroughly depicted net of interactions among ROS, IR, HG, and inflammation, see the review by Luc et al. [79].
2.4. Dyslipidemia and Oxidative Stress
Dyslipidemia in MetS is a state with elevated level of plasma TG associated with increased level of very low-density lipoprotein (VLDL), small, dense LDL (sdLDL-C), FFAs and low HDL cholesterol level that promotes the development of atherosclerosis [80]. Indeed, oxidatively modified LDL (ox-LDL) is an important player in inducing the process of atherosclerosis as it affects expression of adhesive molecules, cytokines and growth factors and changes function of important vasoactive molecules such as NO, angiotensin II (Ang II) or endothelin 1 (ET 1) [81][82]. Interestingly, treatment of MetS patients with rosuvastatin causes beneficial effect not only on levels of ox-LDL, HDL, and inflammatory markers, but also ameliorates total antioxidant capability [83]. Furthermore, high cholesterol level is a promoter of OxS in endothelial cells [84]. In an extensive review, Spahis et al. enumerated several types of connection between dyslipidemia and OxS in MetS. For instance, elevation of O2−• by NADPH occurring upon obesity/HT/hypertriglyceridemia, as well as lowered level of bilirubin in MetS (a protective agent against LDL oxidation), magnitude of LDL oxidation being dependent on waist circumference (visceral adiposity) or ox-LDL affecting mitochondrial functionality [14]. Hyperlipidemia triggers elevation of ROS and proinflammatory cytokines which may be causative factors for lipotoxicity, being predominantly known to be caused by increased rate of lipolysis and repressed synthesis of TG in obesity-affected, insulin-resistant WAT, triggering increased levels of circulating FFA and accumulation of lipids in non-adipose organs (e.g., liver, pancreas, muscles) [85]. For instance, in the recent study by Feillet-Coudray et al., a high-fat high-fructose diet in Wistar rats led to excessive weight gain along with glucose intolerance and hepatic steatosis with elevation of ceramides and DAG (lipotoxicity indicators). Moreover, there was an increase in hepatic NOX activity and protein level of IL-6 along with decrease of total GSSG and GSH as well as activity of SOD and GPx. As these phenomena were associated with moderately marked OxS and inflammation, metabolic alterations were rather suggested to be the trigger, yet, not the cause of OxS [86]. Interestingly, FFAs were reported to be capable of activating renin-angiotensin system (RAS) in mice adipocytes (3T3L1) by TLR4/NF-κB pathway [87]. In fat cells, RAS is connected with the impairment of preadipocytes differentiation, promotion of lipolysis, along with OxS and inflammation.
2.5. Hypertension and Oxidative Stress
There are numerous studies which reported persistently increased ROS levels in HT, along with vast improvement upon antioxidative treatment [88][89][90][91][92][93][94]. HT is connected with vascular remodeling, increased vasoconstriction and arterial stiffness, activation of immune cells, renal dysfunction, and excitation of sympathetic nervous system. Above phenomena are inseparably connected with endothelial dysfunction, LPO, inflammation, fibrosis and more, thus favoring the notion that OxS constitutes a common molecular phenomenon in multifactorial pathogenesis of HT [95]. Cardiovascular cells generate ROS due to action of several major enzymes: NOX, XOR, ERO, and uncoupled NOS. While NOX-mediated ROS production is a prevailing one in HT, more and more pieces of evidence suggest its crosstalk with mitochondrial and ER-specific ROS due to phenomenon of ROS-induced ROS release (RIRR) [89][95][96]. Specifically, in the recent review by Touyz et al., OxS was shown to be connected with HT via several ways [89]. Firstly, prohypertensive factors such as salt, aldosterone, Ang II, and ET-1 activate NOX enzymes and ROS production, being a trigger for mitochondrial and ER-located ROS formation, all of which are interconnected with inflammation and immune activation, and consequently, HT. Here, it is worthwhile to enumerate the increased expression of proinflammatory factors (e.g., TNF-α, IL-1/6), adhesion molecules, and activation of signaling via proinflammatory pathways (e.g., NF-κB, JNK). Secondly, elevation of vascular ROS elicits activation of Ca2+ channels and further activation of Ca2+-sensitive NOX enzymes. Thirdly, Ang II- and ET-1-dependent signaling via their G-coupled receptors promotes transactivation of growth factor receptors (e.g., insulin-like growth factor 1 (IGF-1R), platelet-derived growth factor receptor (PDGFR)) through various mechanisms, triggering activation of signaling via PI3K/AKT and MAPK pathways [89]. Moreover, under physiological conditions, eNOS produces NO, a molecule of critical importance for vasorelaxation. Under NOX-initiated OxS, eNOS produces superoxide, rather than NO, being called uncoupled eNOS and contributing to a sustained increase of ROS levels [97]. Finally, as thoroughly described by Spahis et al., the accumulation of ox-LDL in vascular endothelium is a source of mitochondria-derived OxS in HT [14].
References
- International Diabetes Federation. IDF Diabetes Atlas, 9th edition; International Diabetes Federation: Brussels, Belgium, 2019
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12, doi:10.1007/s11906-018-0812-z.
- Alberti, K.G.M.M.; Zimmet, P.; Shaw, J. The metabolic syndrome--a new worldwide definition. Lancet 2005, 366, 1059–1062, doi:10.1016/S0140-6736(05)67402-8.
- Alberti, K.G.M.M.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.-C.; James, W.P.T.; Loria, C.M.; Smith, S.C.J. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International. Circulation 2009, 120, 1640–1645, doi:10.1161/CIRCULATIONAHA.109.192644.
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 8267234, doi:10.1155/2019/8267234.
- Yan, L.-J. Pathogenesis of chronic hyperglycemia: From reductive stress to oxidative stress. J. Diabetes Res. 2014, 2014, 137919, doi:10.1155/2014/137919.
- Hecker, M.; Wagner, A.H. Role of protein carbonylation in diabetes. J. Inherit. Metab. Dis. 2018, 41, 29–38, doi:10.1007/s10545-017-0104-9.
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2’ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Heal. Part C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 120–139, doi:10.1080/10590500902885684.
- Baba, S.P.; Bhatnagar, A. Role of thiols in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 133–139, doi:10.1016/j.cotox.2018.03.005.
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.-R.; Harrison, D.G.; Bhatnagar, A. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 119, e39-75, doi:10.1161/RES.0000000000000110.
- Matsuzawa, Y.; Funahashi, T.; Nakamura, T. The concept of metabolic syndrome: Contribution of visceral fat accumulation and its molecular mechanism. J. Atheroscler. Thromb. 2011, 18, 629–639, doi:10.5551/jat.7922.
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.M.; Cahill, P.A. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants 2017, 6, doi:10.3390/antiox6040090.
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alexandria J. Med. 2018, 54, 287–293, doi:10.1016/j.ajme.2017.09.001.
- Spahis, S.; Borys, J.-M.; Levy, E. Metabolic Syndrome as a Multifaceted Risk Factor for Oxidative Stress. Antioxid. Redox Signal. 2017, 26, 445–461, doi:10.1089/ars.2016.6756.
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004, 114, 1752–1761, doi:10.1172/JCI21625.
- Carrier, A. Metabolic Syndrome and Oxidative Stress: A Complex Relationship. Antioxid. Redox Signal. 2017, 26, 429–431.
- Le, N.-A. Postprandial Triglycerides, Oxidative Stress, and Inflammation. In Apolipoproteins, Triglycerides and Cholesterol; Waisundara, V.Y., Jovandaric, M.Z., Eds.; IntechOpen: Rijeka, Croatia, 2020.
- Armutcu, F.; Ataymen, M.; Atmaca, H.; Gurel, A. Oxidative stress markers, C-reactive protein and heat shock protein 70 levels in subjects with metabolic syndrome. Clin. Chem. Lab. Med. 2008, 46, 785–790, doi:10.1515/CCLM.2008.166.
- Zelzer, S.; Fuchs, N.; Almer, G.; Raggam, R.B.; Prüller, F.; Truschnig-Wilders, M.; Schnedl, W.; Horejsi, R.; Möller, R.; Weghuber, D.; et al. High density lipoprotein cholesterol level is a robust predictor of lipid peroxidation irrespective of gender, age, obesity, and inflammatory or metabolic biomarkers. Clin. Chim. Acta. 2011, 412, 1345–1349, doi:10.1016/j.cca.2011.03.031.
- Simão, A.N.C.; Lozovoy, M.A.B.; Simão, T.N.C.; Venturini, D.; Barbosa, D.S.; Dichi, J.B.; Matsuo, T.; Cecchini, R.; Dichi, I. Immunological and biochemical parameters of patients with metabolic syndrome and the participation of oxidative and nitroactive stress. Brazilian J. Med. Biol. Res. Rev. Bras. Pesqui. Med. Biol. 2011, 44, 707–712, doi:10.1590/s0100-879x2011007500069.
- Da Fonseca, L.J.S.; Nunes-Souza, V.; Guedes, G. da S.; Schettino-Silva, G.; Mota-Gomes, M.A.; Rabelo, L.A. Oxidative status imbalance in patients with metabolic syndrome: Role of the myeloperoxidase/hydrogen peroxide axis. Oxid. Med. Cell. Longev. 2014, 2014, 898501, doi:10.1155/2014/898501.
- Fujita, K.; Nishizawa, H.; Funahashi, T.; Shimomura, I.; Shimabukuro, M. Systemic oxidative stress is associated with visceral fat accumulation and the metabolic syndrome. Circ. J. 2006, 70, 1437–1442, doi:10.1253/circj.70.1437.
- Caimi, G.; Hopps, E.; Montana, M.; Noto, D.; Canino, B.; Lo Presti, R.; Averna, M.R. Evaluation of nitric oxide metabolites in a group of subjects with metabolic syndrome. Diabetes Metab. Syndr. 2012, 6, 132–135, doi:10.1016/j.dsx.2012.09.012.
- Caimi, G.; Hopps, E.; Noto, D.; Canino, B.; Montana, M.; Lucido, D.; Lo Presti, R.; Averna, M.R. Protein oxidation in a group of subjects with metabolic syndrome. Diabetes Metab. Syndr. 2013, 7, 38–41, doi:10.1016/j.dsx.2013.02.013.
- Caimi, G.; Lo Presti, R.; Montana, M.; Noto, D.; Canino, B.; Averna, M.R.; Hopps, E. Lipid peroxidation, nitric oxide metabolites, and their ratio in a group of subjects with metabolic syndrome. Oxid. Med. Cell. Longev. 2014, 2014, 824756, doi:10.1155/2014/824756.
- Korkmaz, G.G.; Altınoglu, E.; Civelek, S.; Sozer, V.; Erdenen, F.; Tabak, O.; Uzun, H. The association of oxidative stress markers with conventional risk factors in the metabolic syndrome. Metabolism 2013, 62, 828–835, doi:10.1016/j.metabol.2013.01.002.
- Zurawska-Płaksej, E.; Grzebyk, E.; Marciniak, D.; Szymańska-Chabowska, A.; Piwowar, A. Oxidatively modified forms of albumin in patients with risk factors of metabolic syndrome. J. Endocrinol. Invest. 2014, 37, 819–827, doi:10.1007/s40618-014-0111-8.
- Venturini, D.; Simão, A.N.C.; Dichi, I. Advanced oxidation protein products are more related to metabolic syndrome components than biomarkers of lipid peroxidation. Nutr. Res. 2015, 35, 759–765, doi:10.1016/j.nutres.2015.06.013.
- Jialal, I.; Devaraj, S.; Adams-Huet, B.; Chen, X.; Kaur, H. Increased cellular and circulating biomarkers of oxidative stress in nascent metabolic syndrome. J. Clin. Endocrinol. Metab. 2012, 97, E1844–E1850, doi:10.1210/jc.2012-2498.
- Yubero-Serrano, E.M.; Delgado-Lista, J.; Peña-Orihuela, P.; Perez-Martinez, P.; Fuentes, F.; Marin, C.; Tunez, I.; Tinahones, F.J.; Perez-Jimenez, F.; Roche, H.M.; et al. Oxidative stress is associated with the number of components of metabolic syndrome: Lipgene study. Exp. Mol. Med. 2013, 45, e28, doi:10.1038/emm.2013.53.
- Włodarski, A.; Strycharz, J.; Wróblewski, A.; Kasznicki, J., Drzewoski, J.; Śliwińska, A. The Role of microRNAs in Metabolic Syndrome-Related Oxidative Stress. Int. J. Mol. Sci. 2020, doi:10.3390/ijms21186902
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139, doi:10.1016/j.mce.2009.08.018.
- Grant, R.W.; Dixit, V.D. Adipose tissue as an immunological organ. Obesity 2015, 23, 512–518, doi:10.1002/oby.21003.
- Wróblewski, A.; Strycharz, J.; Świderska, E.; Drewniak, K.; Drzewoski, J.; Szemraj, J.; Kasznicki, J.; Śliwińska, A. Molecular Insight into the Interaction between Epigenetics and Leptin in Metabolic Disorders. Nutrients 2019, 11, doi:10.3390/nu11081872.
- Adamczak, M.; Wiecek, A. The adipose tissue as an endocrine organ. Semin. Nephrol. 2013, 33, 2–13, doi:10.1016/j.semnephrol.2012.12.008.
- Ouchi, N.; Ohashi, K.; Shibata, R.; Murohara, T. Adipocytokines and obesity-linked disorders. Nagoya J. Med. Sci. 2012, 74, 19–30.
- Benrick, A.; Chanclón, B.; Micallef, P.; Wu, Y.; Hadi, L.; Shelton, J.M.; Stener-Victorin, E.; Wernstedt Asterholm, I. Adiponectin protects against development of metabolic disturbances in a PCOS mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, E7187–E7196, doi:10.1073/pnas.1708854114.
- Esfahani, M.; Movahedian, A.; Baranchi, M.; Goodarzi, M.T. Adiponectin: An adipokine with protective features against metabolic syndrome. Iran. J. Basic Med. Sci. 2015, 18, 430–442.
- Janochova, K.; Haluzik, M.; Buzga, M. Visceral fat and insulin resistance—What we know? Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech. Repub. 2019, 163, 19–27, doi:10.5507/bp.2018.062.
- Strycharz, J.; Drzewoski, J.; Szemraj, J.; Sliwinska, A. Is p53 Involved in Tissue-Specific Insulin Resistance Formation? Oxid. Med. Cell. Longev. 2017, 2017, 9270549, doi:10.1155/2017/9270549.
- Xu, L.; Kitade, H.; Ni, Y.; Ota, T. Roles of chemokines and chemokine receptors in obesity-associated insulin resistance and nonalcoholic fatty liver disease. Biomolecules 2015, 5, 1563–1579.
- Huber, J.; Kiefer, F.W.; Zeyda, M.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; Zlabinger, G.J.; Stulnig, T.M. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J. Clin. Endocrinol. Metab. 2008, doi:10.1210/jc.2007-2630.
- Zand, H.; Morshedzadeh, N.; Naghashian, F. Signaling pathways linking inflammation to insulin resistance. Diabetes Metab. Syndr. 2017, 11 (Suppl. 1) S307–S309, doi:10.1016/j.dsx.2017.03.006.
- Kwon, H.; Pessin, J.E.; Adipokines, Inflammation, and Insulin Resistance in Obesity. In Textbook of Energy Balance, Neuropeptide Hormones, and Neuroendocrine Function; Nillni, E.A., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 225–252. ISBN 978-3-319-89506-2.
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023, doi:10.1038/sigtrans.2017.23.
- Ohashi, K.; Shibata, R.; Murohara, T.; Ouchi, N. Role of anti-inflammatory adipokines in obesity-related diseases. Trends Endocrinol. Metab. 2014, 25, 348–355, doi:10.1016/j.tem.2014.03.009.
- Pan, X.; Kaminga, A.C.; Wen, S.W.; Acheampong, K.; Liu, A. Omentin-1 in diabetes mellitus: A systematic review and meta-analysis. PLoS ONE 2019, 14, e0226292, doi:10.1371/journal.pone.0226292.
- Engin, A.B. What Is Lipotoxicity? Adv. Exp. Med. Biol. 2017, 960, 197–220, doi:10.1007/978-3-319-48382-5_8.
- Hauck, A.K.; Huang, Y.; Hertzel, A. V.; Bernlohr, D.A. Adipose oxidative stress and protein carbonylation. J. Biol. Chem. 2019, 294, 1083–1088, doi:10.1074/jbc.R118.003214.
- Le Lay, S.; Simard, G.; Martinez, M.C.; Andriantsitohaina, R. Oxidative stress and metabolic pathologies: From an adipocentric point of view. Oxid. Med. Cell. Longev. 2014, 2014, 908539, doi:10.1155/2014/908539.
- Wang, C.-H.; Wang, C.-C.; Huang, H.-C.; Wei, Y.-H. Mitochondrial dysfunction leads to impairment of insulin sensitivity and adiponectin secretion in adipocytes. FEBS J. 2013, 280, 1039–1050, doi:10.1111/febs.12096.
- Ortega, S.P.; Chouchani, E.T.; Boudina, S. Stress turns on the heat: Regulation of mitochondrial biogenesis and UCP1 by ROS in adipocytes. Adipocyte 2017, 6, 56–61, doi:10.1080/21623945.2016.1273298.
- Levy-Marchal, C.; Arslanian, S.; Cutfield, W.; Sinaiko, A.; Druet, C.; Marcovecchio, M.L.; Chiarelli, F. Insulin resistance in children: Consensus, perspective, and future directions. J. Clin. Endocrinol. Metab. 2010, 95, 5189–5198, doi:10.1210/jc.2010-1047.
- Onyango, A.N. Cellular Stresses and Stress Responses in the Pathogenesis of Insulin Resistance. Oxid. Med. Cell. Longev. 2018, 2018, 4321714, doi:10.1155/2018/4321714.
- Świderska, E., Strycharz, J., Wróblewski, A., Szemraj, J., Drzewoski, J., Śliwińska, A. Role of PI3K/AKT Pathway in Insulin-Mediated Glucose Uptake. In Blood Glucose Levels; IntechOpen: London, UK, 2018.
- Kolka, C.M.; Bergman, R.N. The endothelium in diabetes: Its role in insulin access and diabetic complications. Rev. Endocr. Metab. Disord. 2013, 14, 13–19, doi:10.1007/s11154-012-9233-5.
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1-R10, doi:10.1530/JME-11-0022.
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pract. 2011, 93 (Suppl. 1) S52–S59, doi:10.1016/S0168-8227(11)70014-6.
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Invest. 2016, 126, 12–22, doi:10.1172/JCI77812.
- Esteves, J.V.; Enguita, F.J.; Machado, U.F. MicroRNAs-Mediated Regulation of Skeletal Muscle GLUT4 Expression and Translocation in Insulin Resistance. J. Diabetes Res. 2017, 2017, 7267910, doi:10.1155/2017/7267910.
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161, doi:10.1002/jcp.27603.
- Chen, Y.; Huang, L.; Qi, X.; Chen, C. Insulin Receptor Trafficking: Consequences for Insulin Sensitivity and Diabetes. Int. J. Mol. Sci. 2019, 20, doi:10.3390/ijms20205007.
- Li, Y.Z.; Di Cristofano, A.; Woo, M. Metabolic Role of PTEN in Insulin Signaling and Resistance. Cold Spring Harb. Perspect. Med. 2020, 10, doi:10.1101/cshperspect.a036137.
- Ma, J.; Nakagawa, Y.; Kojima, I.; Shibata, H. Prolonged insulin stimulation down-regulates GLUT4 through oxidative stress-mediated retromer inhibition by a protein kinase CK2-dependent mechanism in 3T3-L1 adipocytes. J. Biol. Chem. 2014, 289, 133–142, doi:10.1074/jbc.M113.533240.
- Yazıcı, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304, doi:10.1007/978-3-319-48382-5_12.
- Pereira, S.; Park, E.; Mori, Y.; Haber, C.A.; Han, P.; Uchida, T.; Stavar, L.; Oprescu, A.I.; Koulajian, K.; Ivovic, A.; et al. FFA-induced hepatic insulin resistance in vivo is mediated by PKCδ, NADPH oxidase, and oxidative stress. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E34–E46, doi:10.1152/ajpendo.00436.2013.
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871, doi:10.1016/j.cell.2012.02.017.
- Tangvarasittichai, S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J. Diabetes 2015, 6, 456–480, doi:10.4239/wjd.v6.i3.456.
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262, doi:10.1016/j.bj.2017.06.007.
- Dokken, B.B.; Saengsirisuwan, V.; Kim, J.S.; Teachey, M.K.; Henriksen, E.J. Oxidative stress-induced insulin resistance in rat skeletal muscle: Role of glycogen synthase kinase-3. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E615–E621, doi:10.1152/ajpendo.00578.2007.
- Ighodaro, O.M. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed. Pharmacother. 2018, 108, 656–662, doi:10.1016/j.biopha.2018.09.058.
- Dozza, B.; Smith, M.A.; Perry, G.; Tabaton, M.; Strocchi, P. Regulation of glycogen synthase kinase-3beta by products of lipid peroxidation in human neuroblastoma cells. J. Neurochem. 2004, 89, 1224–1232, doi:10.1111/j.1471-4159.2004.02413.x.
- Grimsrud, P.A.; Picklo, M.J.; Griffin, T.J.; Bernlohr, D.A. Carbonylation of adipose proteins in obesity and insulin resistance: Identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol. Cell. Proteomics 2007, 6, 624–637, doi:10.1074/mcp.M600120-MCP200.
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945, doi:10.2337/diabetes.49.11.1939.
- Gabbay, K.H.; Merola, L.O.; Field, R.A. Sorbitol pathway: Presence in nerve and cord with substrate accumulation in diabetes. Science 1966, 151, 209–210, doi:10.1126/science.151.3707.209.
- Nigro, C.; Raciti, G.A.; Leone, A.; Fleming, T.H.; Longo, M.; Prevenzano, I.; Fiory, F.; Mirra, P.; D’Esposito, V.; Ulianich, L.; et al. Methylglyoxal impairs endothelial insulin sensitivity both in vitro and in vivo. Diabetologia 2014, 57, 1485–1494, doi:10.1007/s00125-014-3243-7.
- Schober, A. Chemokines in vascular dysfunction and remodeling. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1950–1959, doi:10.1161/ATVBAHA.107.161224.
- Meerwaldt, R.; Links, T.; Zeebregts, C.; Tio, R.; Hillebrands, J.-L.; Smit, A. The clinical relevance of assessing advanced glycation endproducts accumulation in diabetes. Cardiovasc. Diabetol. 2008, 7, 29, doi:10.1186/1475-2840-7-29.
- Luc, K.; Schramm-Luc, A.; Guzik, T.J.; Mikolajczyk, T.P. Oxidative stress and inflammatory markers in prediabetes and diabetes. J. Physiol. Pharmacol. 2019, 70, doi:10.26402/jpp.2019.6.01.
- Blaton, V. How is the Metabolic Syndrome Related to the Dyslipidemia? EJIFCC 2007, 18, 15–22.
- Jabarpour, M.; Rashtchizadeh, N.; Argani, H.; Ghorbanihaghjo, A.; Ranjbarzadhag, M.; Sanajou, D.; Panah, F.; Alirezaei, A. The impact of dyslipidemia and oxidative stress on vasoactive mediators in patients with renal dysfunction. Int. Urol. Nephrol. 2019, 51, 2235–2242, doi:10.1007/s11255-019-02319-7.
- Rizzo, M.; Kotur-Stevuljevic, J.; Berneis, K.; Spinas, G.; Rini, G.B.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V.; Vekic, J. Atherogenic dyslipidemia and oxidative stress: A new look. Transl. Res. 2009, 153, 217–223, doi:10.1016/j.trsl.2009.01.008.
- Bostan, C.; Yildiz, A.; Ozkan, A.A.; Uzunhasan, I.; Kaya, A.; Yigit, Z. Beneficial effects of rosuvastatin treatment in patients with metabolic syndrome. Angiology 2015, 66, 122–127, doi:10.1177/0003319714522107.
- Razavi, S.-M.; Gholamin, S.; Eskandari, A.; Mohsenian, N.; Ghorbanihaghjo, A.; Delazar, A.; Rashtchizadeh, N.; Keshtkar-Jahromi, M.; Argani, H. Red grape seed extract improves lipid profiles and decreases oxidized low-density lipoprotein in patients with mild hyperlipidemia. J. Med. Food 2013, 16, 255–258, doi:10.1089/jmf.2012.2408.
- Manzoni, A.G.; Passos, D.F.; Leitemperger, J.W.; Storck, T.R.; Doleski, P.H.; Jantsch, M.H.; Loro, V.L.; Leal, D.B.R. Hyperlipidemia-induced lipotoxicity and immune activation in rats are prevented by curcumin and rutin. Int. Immunopharmacol. 2020, 81, 106217, doi:10.1016/j.intimp.2020.106217.
- Feillet-Coudray, C.; Fouret, G.; Vigor, C.; Bonafos, B.; Jover, B.; Blachnio-Zabielska, A.; Rieusset, J.; Casas, F.; Gaillet, S.; Landrier, J.F.; et al. Long-Term Measures of Dyslipidemia, Inflammation, and Oxidative Stress in Rats Fed a High-Fat/High-Fructose Diet. Lipids 2019, 54, 81–97, doi:10.1002/lipd.12128.
- Sun, J.; Luo, J.; Ruan, Y.; Xiu, L.; Fang, B.; Zhang, H.; Wang, M.; Chen, H. Free Fatty Acids Activate Renin-Angiotensin System in 3T3-L1 Adipocytes through Nuclear Factor-kappa B Pathway. J. Diabetes Res. 2016, 2016, 1587594, doi:10.1155/2016/1587594.
- Levy, E.; Spahis, S.; Bigras, J.-L.; Delvin, E.; Borys, J.-M. The Epigenetic Machinery in Vascular Dysfunction and Hypertension. Curr. Hypertens. Rep. 2017, 19, 52, doi:10.1007/s11906-017-0745-y.
- Touyz, R.M.; Rios, F.J.; Alves-Lopes, R.; Neves, K.B.; Camargo, L.L.; Montezano, A.C. Oxidative Stress: A Unifying Paradigm in Hypertension. Can. J. Cardiol. 2020, 36, 659–670, doi:10.1016/j.cjca.2020.02.081.
- Coats, A.; Jain, S. Protective effects of nebivolol from oxidative stress to prevent hypertension-related target organ damage. J. Hum. Hypertens. 2017, 31, 376–381, doi:10.1038/jhh.2017.8.
- Welch, W.J.; Mendonca, M.; Blau, J.; Karber, A.; Dennehy, K.; Patel, K.; Lao, Y.S.; José, P.A.; Wilcox, C.S. Antihypertensive response to prolonged tempol in the spontaneously hypertensive rat. Kidney Int. 2005, doi:10.1111/j.1523-1755.2005.00392.x.
- Adlakha, Y.K.; Khanna, S.; Singh, R.; Singh, V.P.; Agrawal, A.; Saini, N. Pro-apoptotic miRNA-128-2 modulates ABCA1, ABCG1 and RXRα expression and cholesterol homeostasis. Cell Death Dis. 2013, 4, e780, doi:10.1038/cddis.2013.301.
- Dikalova, A.E.; Pandey, A.; Xiao, L.; Arslanbaeva, L.; Sidorova, T.; Lopez, M.G.; Billings, F.T.; Verdin, E.; Auwerx, J.; Harrison, D.G.; et al. Mitochondrial deacetylase SIRT3 reduces vascular dysfunction and hypertension while SIRT3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circ. Res. 2020, doi:10.1161/CIRCRESAHA.119.315767.
- Li, G.; Wang, X.; Yang, H.; Zhang, P.; Wu, F.; Li, Y.; Zhou, Y.; Zhang, X.; Ma, H.; Zhang, W.; et al. α-Linolenic acid but not linolenic acid protects against hypertension: Critical role of SIRT3 and autophagic flux. Cell Death Dis. 2020, doi:10.1038/s41419-020-2277-7.
- Gong, Y.Y.; Luo, J.Y.; Wang, L.; Huang, Y. MicroRNAs Regulating Reactive Oxygen Species in Cardiovascular Diseases. Antioxid. Redox Signal. 2018, 29, 1092–1107.
- Zinkevich, N.S.; Gutterman, D.D. ROS-induced ROS release in vascular biology: Redox-redox signaling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H647–H653, doi:10.1152/ajpheart.01271.2010.
- Li, Q.; Yon, J.; Cai, H.; Angeles, C.L.; Angeles, L. Mechanisms and Consequences of eNOS Dysfunction in Hypertension. J. Hypertens. 2016, doi:10.1097/HJH.0000000000000587.

