 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Furong TANG | -- | 1415 | 2022-04-22 03:37:10 | | | |
| 2 | Catherine Yang | Meta information modification | 1415 | 2022-04-22 04:13:10 | | |
Video Upload Options
Pairwise sequence alignment is the basis of multiple sequence alignment and mainly divided into local alignment and global alignment. The former is to find and align the similar local region, and the latter is end-to-end alignment. A commonly used global alignment algorithm is the Needleman–Wunsch algorithm, which has become the basic algorithm that is used in many types of multiple sequence alignment software. The algorithm usually consists of two steps: one is calculating the states of the dynamic programming matrix; and the other is tracking back from the final state to the initial state of the dynamic programming matrix to obtain the solution of alignment. Time and space complexity of pairwise sequence alignment algorithms based on dynamic programming is O(l1l2), where l1 and l2 are the lengths of the two sequences to be aligned. Such overheads are acceptable for short sequences but not for sequences with more than several thousand sites. As a space-saving strategy of the dynamic programming algorithm, the Hirschberg algorithm is able to complete alignment by the space complexity of O(l) without any sacrifice of quality.
1. Divide and Conquer
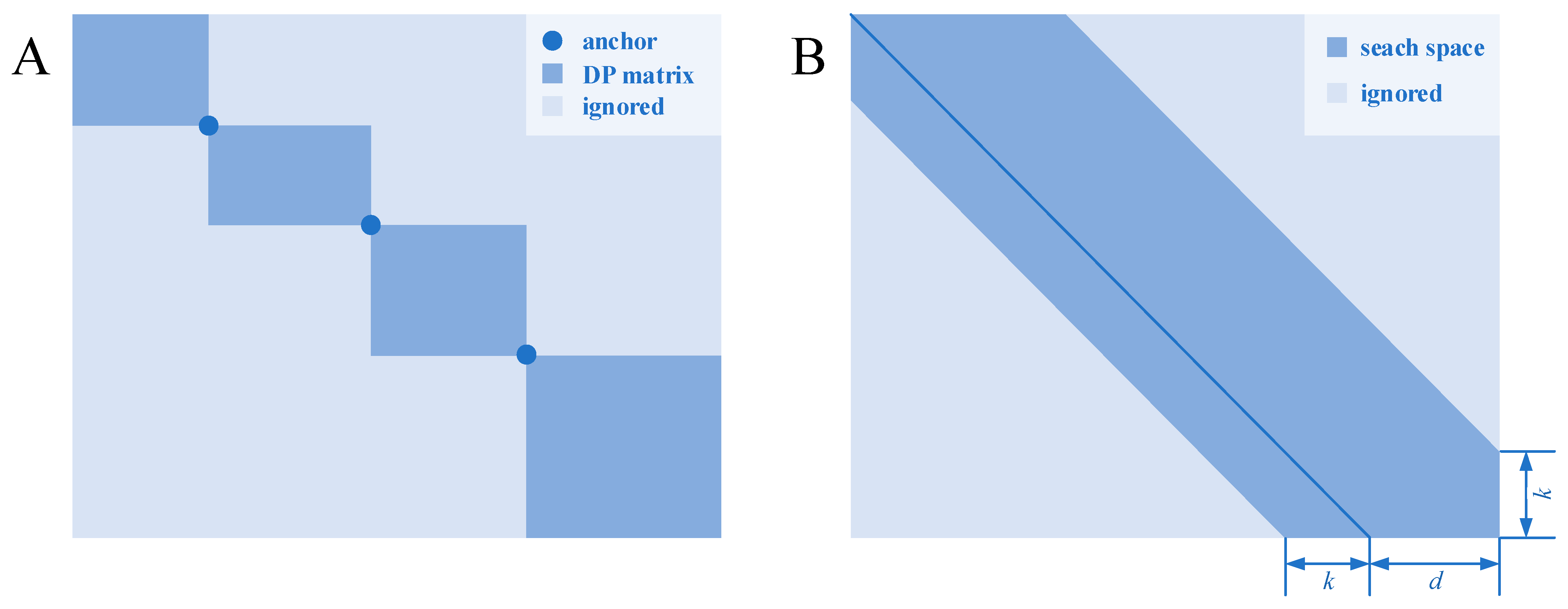
2. Bounded Dynamic Programming
3. Scoring System of Pairwise Sequence Alignment
References
- Lipman, D.J.; Pearson, W.R. Rapid and Sensitive Protein Similarity Searches. Science 1985, 227, 1435–1441.
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448.
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410.
- Karp, R.M.; Rabin, M.O. Efficient randomized pattern-matching algorithms. IBM J. Res. Dev. 1987, 31, 249–260.
- Delcher, A.L.; Kasif, S.; Fleischmann, R.D.; Peterson, J.; White, O.; Salzberg, S.L. Alignment of whole genomes. Nucleic Acids Res. 1999, 27, 2369–2376.
- Marçais, G.; Delcher, A.L.; Phillippy, A.; Coston, R.; Salzberg, S.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLOS Comput. Biol. 2018, 14, e1005944.
- Weiner, P. Linear pattern matching algorithms. In Proceedings of the 14th Annual Symposium on Switching and Automata Theory (Swat 1973), Iowa City, IA, USA, 15–17 October 1973; pp. 1–11.
- Manber, U.; Myers, G. Suffix Arrays: A New Method for On-Line String Searches. SIAM J. Comput. 1993, 22, 935–948.
- Ferragina, P.; Manzini, G. Opportunistic data structures with applications. In Proceedings of the 41st Annual Symposium on Foundations of Computer Science, Redondo Beach, CA, USA, 12–14 November 2000; pp. 390–398.
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100.
- Moshiri, N. ViralMSA: Massively scalable reference-guided multiple sequence alignment of viral genomes. Bioinformatics 2021, 37, 714–716.
- Kazutaka, K.; Misakwa, K.; Kei-ichi, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066.
- Naznooshsadat, E.; Elham, P.; Ali, S.-Z.; Etminan, N.; Parvinnia, E.; Sharifi-Zarchi, A. FAME: Fast and memory efficient multiple sequences alignment tool through compatible chain of roots. Bioinformatics 2020, 36, 3662–3668.
- Liu, H.; Zou, Q.; Xu, Y. A novel fast multiple nucleotide sequence alignment method based on FM-index. Brief. Bioinform. 2022, 23, bbab519.
- Smirnov, V.; Warnow, T. MAGUS: Multiple sequence Alignment using Graph clUStering. Bioinformatics 2021, 37, 1666–1672.
- Edgar, R.C. MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping. bioRxiv 2021.
- Spouge, J.L. Speeding up Dynamic Programming Algorithms for Finding Optimal Lattice Paths. SIAM J. Appl. Math. 1989, 49, 1552–1566.
- Korf, R.E. Depth-first iterative-deepening: An optimal admissible tree search. Artif. Intell. 1985, 27, 97–109.
- Ranwez, V.; Harispe, S.; Delsuc, F.; Douzery, E.J.P. MACSE: Multiple Alignment of Coding SEquences Accounting for Frameshifts and Stop Codons. PLoS ONE 2011, 6, e22594.
- Li, W.-H.; Wu, C.I.; Luo, C.C. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 1985, 2, 150–174.
- Schwartz, R.M.; Dayhoff, M.O. Matrices for Detecting Distant Relationships. In Atlas of Protein Sequences; National Biomedical Research Foundation: Washington, DC, USA, 1978; pp. 353–359.
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282.
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919.
- Ríos, S.; Fernandez, M.F.; Caltabiano, G.; Campillo, M.; Pardo, L.; Gonzalez, A. GPCRtm: An amino acid substitution matrix for the transmembrane region of class A G Protein-Coupled Receptors. BMC Bioinform. 2015, 16, 206.
- Vingron, M.; Waterman, M.S. Sequence alignment and penalty choice: Review of concepts, case studies and implications. J. Mol. Biol. 1994, 235, 1–12.

