Pairwise sequence alignment is the basis of multiple sequence alignment and mainly divided into local alignment and global alignment. The former is to find and align the similar local region, and the latter is end-to-end alignment. A commonly used global alignment algorithm is the Needleman–Wunsch algorithm, which has become the basic algorithm that is used in many types of multiple sequence alignment software. The algorithm usually consists of two steps: one is calculating the states of the dynamic programming matrix; and the other is tracking back from the final state to the initial state of the dynamic programming matrix to obtain the solution of alignment. Time and space complexity of pairwise sequence alignment algorithms based on dynamic programming is O(l1l2), where l1 and l2 are the lengths of the two sequences to be aligned. Such overheads are acceptable for short sequences but not for sequences with more than several thousand sites. As a space-saving strategy of the dynamic programming algorithm, the Hirschberg algorithm is able to complete alignment by the space complexity of O(l) without any sacrifice of quality.
1. Divide and Conquer
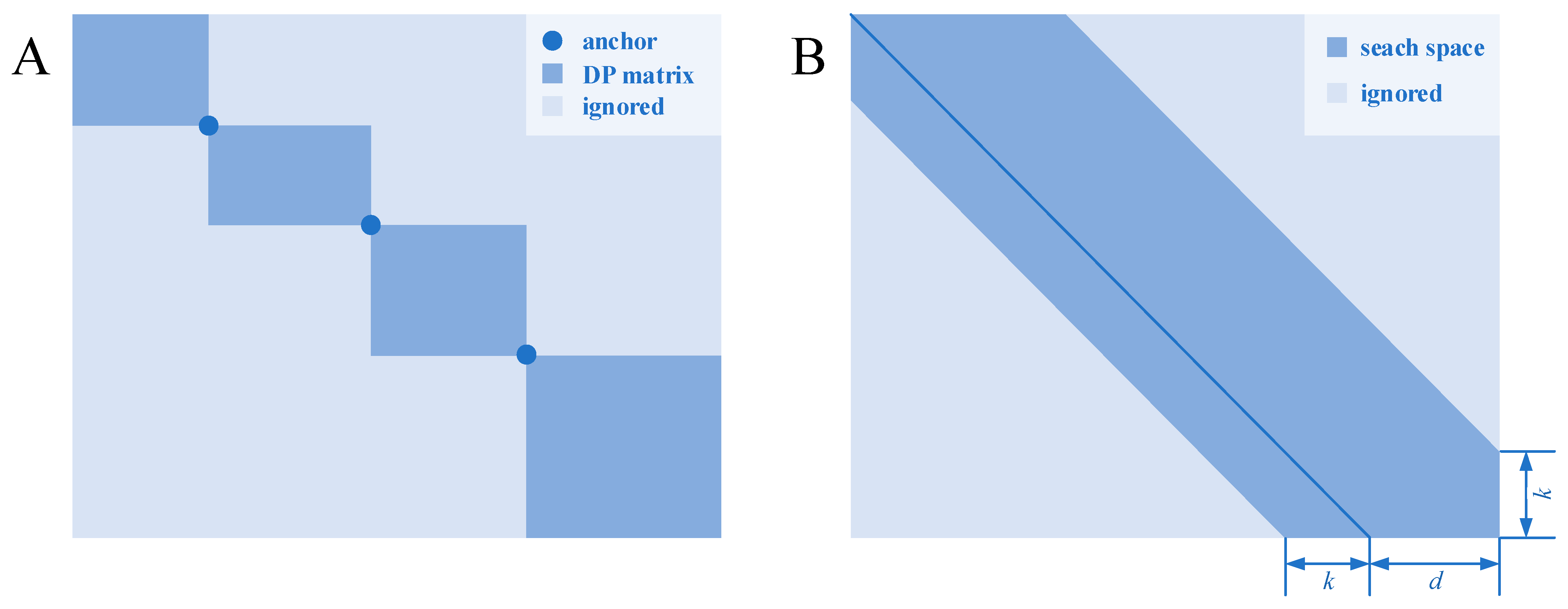
One of the heuristic methods is based on divide and conquer. In such methods, homologous segments (or seeds) are found and used as the “anchors” for the alignment. Each anchor point can divide the dynamic programming matrix into four sub-matrices located at the four corners. Backtracking always goes toward the upper left direction, and these anchors are regarded as the waypoints that the optimal path must pass; therefore, the sub-matrices located at the lower left and upper right are useless and naturally disregarded. When more anchor points distributed throughout the sequences are found, the scale of the dynamic programming matrix can be greatly reduced, thereby reducing the time and space complexity (Figure 1A).
Figure 1. Two heuristic algorithms for pairwise sequence alignment. (A) A dynamic programming matrix, which is separated by several anchors, which is certain to be in the optimal path. (B) A shape-based bounded dynamic programming matrix in which the light blue block is calculation-free because these states are thought to be less likely to be in the optimal path.
Many alignment algorithms are designed to find such seeds to accelerate alignment. FASTA
[21,22][1][2] and BLAST
[23][3] are two of the classic ones. They both search for common substrings in a way similar to the Rabin–Karp algorithm
[24][4]. Moreover, some other software, such as MUMmer
[25[5][6],
26], uses data structures, such as suffix tree
[27][7], suffix array
[28][8], or FM-index
[29][9], to find homologous segments. Recently, Minimap2
[30][10], which also adopts this strategy, is gaining popularity and is used as the basis of ViralMSA
[31][11], which can perform multiple sequence alignment of viral genomes with the help of a reference sequence and is linearly scalable with the number of sequences. MAFFT
[32][12] utilizes fast Fourier transform (FFT) to accelerate the calculation of the correlation between two sequences, which is an indicator of homologous regions of the sequences. These homologous regions can then act as anchors in pairwise sequence alignment. Additionally, it is concluded in Ref
[32][12] that the accuracy of FFT-based methods is almost unaffected by this heuristic approach.
The concept of divide and conquer is also adopted in some multiple sequence alignment software to scale to large datasets, such as FAME
[33][13] and FMAlign
[34][14], which vertically divide the sequences using common seeds among the sequences, and MAGUS
[35][15] and Super5
[36][16], which divide the sequences horizontally into subsets that are small enough to be aligned fast and accurately.
2. Bounded Dynamic Programming
Bounded dynamic programming
[37][17] is based on a heuristic idea: if two sequences have close similarity, then the number of gaps inserted in the sequences during alignment will be relatively small. Therefore, the possible backtracking paths will be close to the diagonal of the dynamic programming matrix for similar sequences. The possible interval can be seen as a strip with certain width parallel to the diagonal (
Figure 1B). The states located in the strip are calculated, while the others are ignored. The width of the strip reflects the trade-off between the alignment accuracy and time consumption: a wide strip means more states needed to be calculated, whereas a narrow strip means that more states could be ignored, which will, however, increase the possibility of missing the optimal path.
Several methods are available for determining the strip range. A basic idea is to use the shape-based division, but this does not fully consider the biological significance and is rarely used. A simple improvement of this method is to set a threshold to filter the states that could be ignored. If the score of one state in the dynamic programming plus the score for the transition from this state to the final state is greater than the threshold, then transitions from this state are allowed. However, this approach requires transition scores to be estimated and a threshold to be set. The transition scores can be conservatively estimated by its upper bound, and the threshold can be generally determined using the iterative method proposed in Ref
[38][18].
3. Scoring System of Pairwise Sequence Alignment
The most critical factor for the quality of a pairwise sequence alignment is the scoring system. It is the basis of the sequence alignment, including multiple sequence alignment, because it determines the direction of the alignment and reflects its quality. Most types of the sequence alignment software aim to obtain good alignment by defining an explicit or implicit objective function for scoring and improving their ability to achieve high score by adjusting alignment strategy. The higher score an alignment can achieve, the higher
wresearche
rs think its accuracy will be in the corresponding scoring system. As an example, a model and the corresponding scoring system for pairwise alignment of nucleotide sequences containing frameshifts and stop codons comprise the main feature of MACSE, a multiple sequence alignment tool that is specific to coding sequences and takes into account frameshifts and stop codons
[39][19].
Generally, the score of a pairwise sequence alignment is the sum of the scores of all aligned pairs. For alignment of two protein sequences, for example, each pair of aligned sites is scored depending on whether a gap is involved, or, if no gaps are involved, whether the two aligned residues are matched or mismatched. When a gap is involved, a gap penalty, which is usually a negative score, is given. Additionally, the score for matched and mismatched amino acid residues is generally determined using a substitution matrix.
The most basic substitution matrix is based on whether the two residues are matched or not. More complex matrices also take into consideration the attributes of nucleotides or amino acid residues. For example, the score of conversion–transversion matrix reflects the difference between conversion and transversion frequency in natural mutations
[40][20]. Protein sequences contain more types of residues than nucleic acid sequences, and therefore, the substitutions and the frequency involved are more complicated. Early amino acid substitution matrix is based on the properties and the codons of amino acids
[41][21]. More recent amino acid substitution matrices rely on the analysis of the substitution frequency of a large number of homologous sequences
[41][21] and aim to reflect the natural probability of substitutions among amino acids at certain evolutionary distances by giving conservative substitutions higher scores. Typical amino acid substitution matrices are percent accepted mutations (PAM)
[41,42][21][22] and BLOcks SUbstitution matrix (BLOSUM)
[43][23]. Although these matrices are widely adopted, there are also matrices designed for specific protein domain, such as GPCRtm, which is summarized from the transmembrane segments of the G-protein-coupled receptor (GPCR) rhodopsin family for the reason that it is not optimal to align sequences with marked compositional biases using the general-purpose matrices
[44][24].
In addition to substitution matrices, gap penalty is also an important part of the scoring system. A simple rule is to assign a fixed negative score when a nucleotide or amino acid residue aligns with a gap. However, this scoring method has some intrinsic limitations, mainly because insertions and deletions (indels) are small-probability events, especially in nucleic acid sequences where indels can cause frameshifts and disrupt all subsequent codons. Once a gap is inserted in an alignment, adjacent gaps are more likely to occur compared with gaps inserted at a distance from the first gap. Therefore, almost all sequence alignment algorithms now use the gap penalty rule based on the number of the gaps successively inserted, and the most typical one is the affine penalty. No optimal solution is universally applicable for the gap penalty, which is referred to as a “black art” requiring constant trial of errors
[45][25].