 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cristina Teixidó | -- | 2974 | 2022-04-21 19:36:45 | | | |
| 2 | Vivi Li | -32 word(s) | 2942 | 2022-04-22 04:58:23 | | |
Video Upload Options
Melanoma develops as a result of several genetic alterations, with UV radiation often acting as a mutagenic risk factor. Deep knowledge of the molecular signaling pathways of different types of melanoma allows better characterization and provides tools for the development of therapies based on the intervention of signals promoted by these cascades. The latest World Health Organization classification acknowledged the specific genetic drivers leading to melanoma and classifies melanocytic lesions into nine distinct categories according to the associate cumulative sun damage (CSD), which correlates with the molecular alterations of tumors. The largest groups are melanomas associated with low-CSD or superficial spreading melanomas, characterized by frequent presentation of the BRAFV600 mutation. High-CSD melanomas include lentigo maligna type and desmoplastic melanomas, which often have a high mutation burden and can harbor NRAS, BRAFnon-V600E, or NF1 mutations. Non-CSD-associated melanomas encompass acral and mucosal melanomas that usually do not show BRAF, NRAS, or NF1 mutations (triple wild-type), but in a subset may have KIT or SF3B1 mutations. To improve survival, these driver alterations can be treated with targeted therapy achieving significant antitumor activity.
1. Introduction
1.1. Epidemiology
2. Molecular Pathways of Melanoma Development
2.1. MAPK Pathway
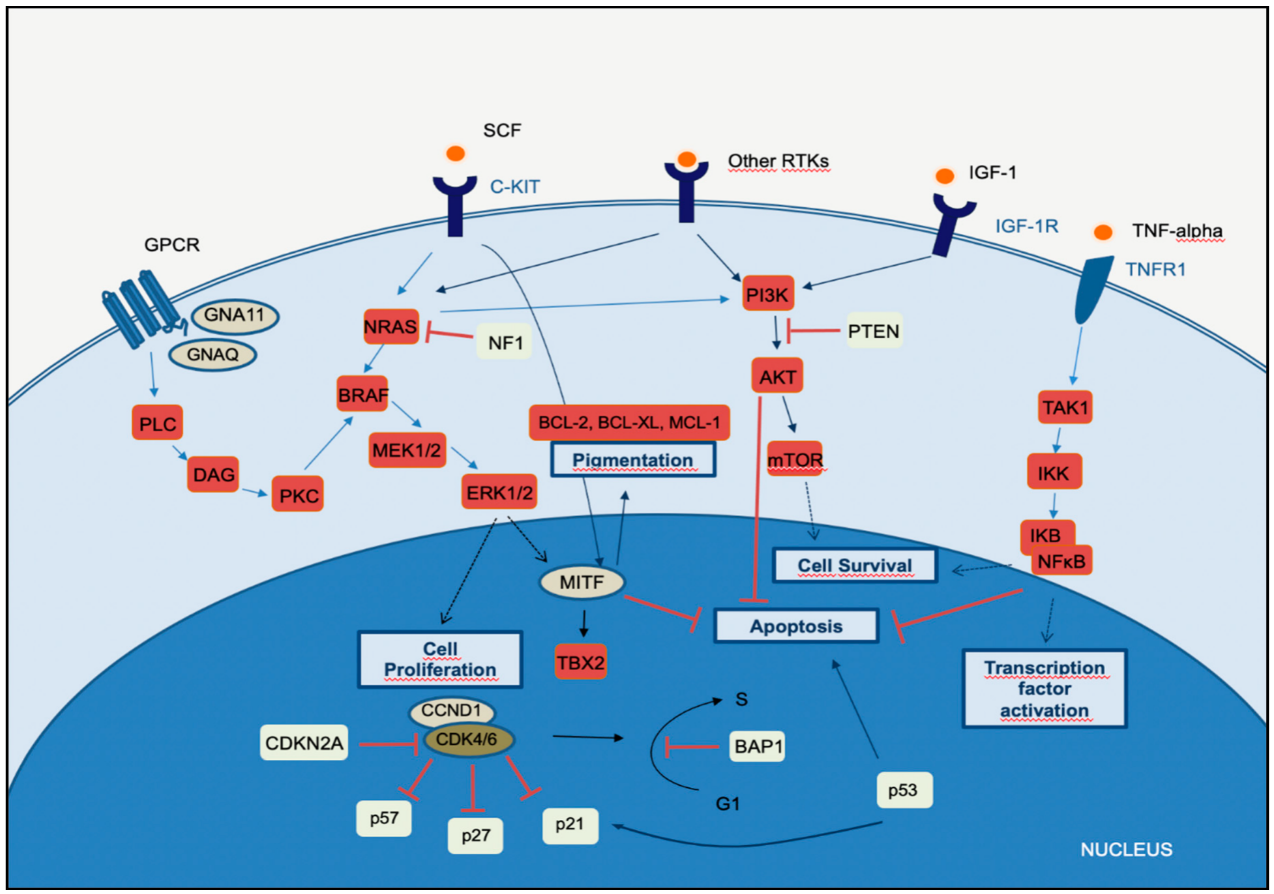
2.2. PI3K-AKT Pathway
2.3. CDKN2A, Cell Cycle, and Apoptosis Regulation
2.4. MITF Pathway
3. The Integration of Histology and Molecular Diagnostics of Melanoma
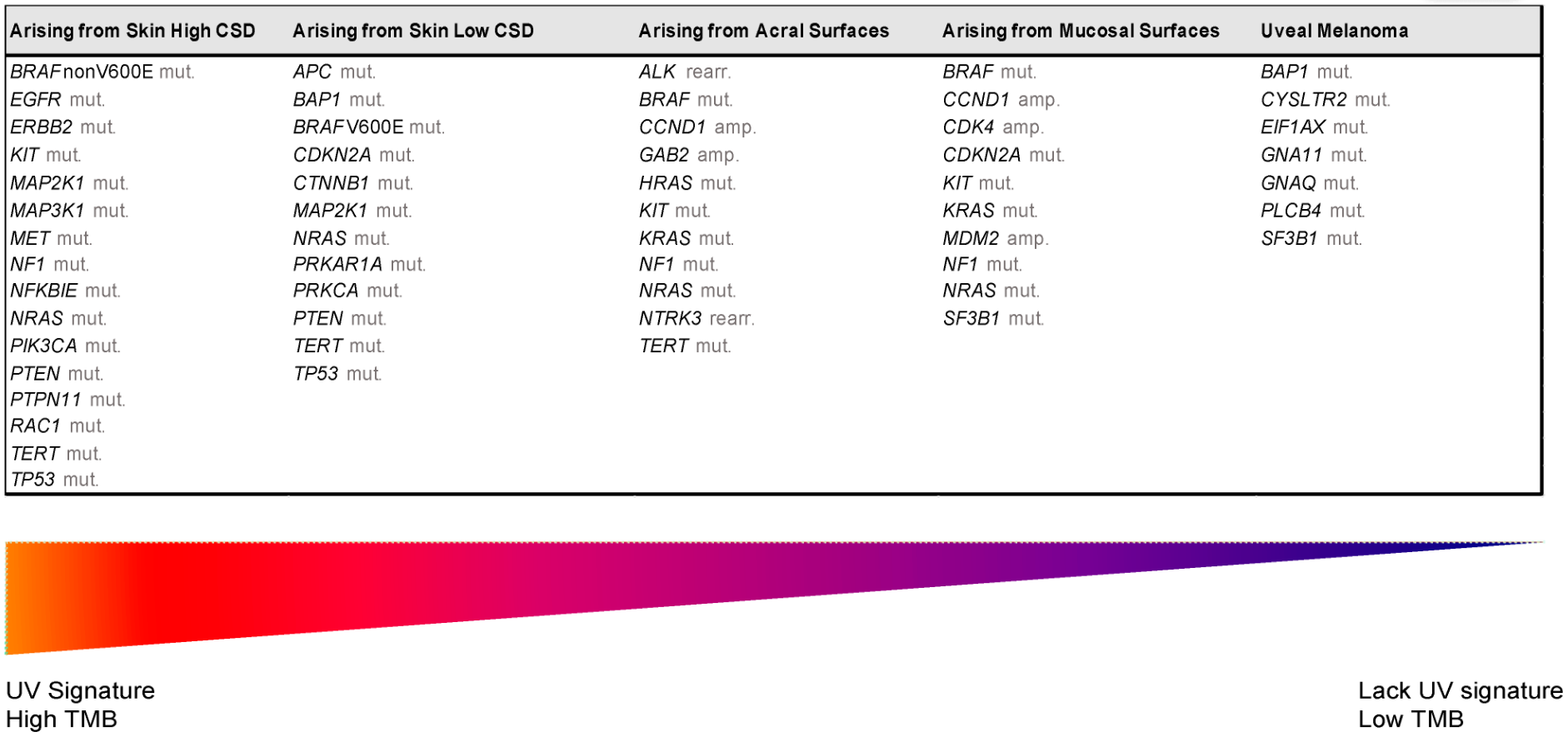
| UV Exposure | Categories | Melanoma Subtype | Key Molecular Genes | |
|---|---|---|---|---|
| Low UV/CSD | I | Superficial spreading melanoma | BRAFV600 mut CDKN2A mut NRAS mut |
TERT mut PTEN mut TP53 mut |
| High UV/CSD | II | Lentigo maligna melanoma | NRAS mut BRAFnon-V600E mut KIT mut TERT mut |
CDKN2A mut PTEN mut TP53 mut |
| III | Desmoplastic melanoma | NF1 mut NFKBIE mut |
NRAS mut PIK3CA mut |
|
| Low or no UV/CSD | IV | Spitz melanoma | ALK rearr NTRK1 rearr NRTK3 rearr |
CDKN2A mut HRAS mut |
| V | Acral melanoma | KIT mut NRAS or BRAF mut ALK rearr NRTK3 rearr |
CDKN2A mut CCND1 amp TERT mut |
|
| VI | Mucosal melanoma | KIT mut NRAS or BRAF mut CDKN2A mut SF3B1 mut |
CCND1 amp CDK4 mut MDM2 amp |
|
| VII | Melanoma in congenital nevus | NRAS mut | BRAFV600E mut | |
| VIII | Melanoma in blue nevus | GNA11 mut GNAQ mut CYSLTR2 mut |
BAP1 mut EIFAX mut SF3B1 mut |
|
| IX | Uveal melanoma | GNA11 mut GNAQ mut CYSLTR2 mut PLCB4 mut |
BAP1 mut EIFAX mut SF3B1 mut |
|
References
- Cress, R.D.; Holly, E.A. Incidence of cutaneous melanoma among non-Hispanic whites, Hispanics, Asians, and blacks: An analysis of california cancer registry data, 1988–1993. Cancer Causes Control. 1997, 8, 246–252.
- Ridky, T.W. Nonmelanoma skin cancer. J. Am. Acad. Dermatol. 2007, 57, 484–501.
- Jhappan, C.; Noonan, F.P.; Merlino, G. Ultraviolet radiation and cutaneous malignant melanoma. Oncogene 2003, 22, 3099–3112.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA: A Cancer J. Clin. 2020, 70, 7–30.
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo (Athens, Greece) 2014, 28, 1005–1011.
- Michielin, O.; van Akkooi, A.C.J.; Ascierto, P.A.; Dummer, R.; Keilholz, U. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1884–1901.
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723.
- Fellner, C. Ipilimumab (yervoy) prolongs survival in advanced melanoma: Serious side effects and a hefty price tag may limit its use. P T 2012, 37, 503–530.
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516.
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714.
- Sample, A.; He, Y.-Y. Mechanisms and prevention of UV-induced melanoma. Photodermatol. Photoimmunol. Photomed. 2018, 34, 13–24.
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248.
- Bevona, C.; Goggins, W.; Quinn, T.; Fullerton, J.; Tsao, H. Cutaneous melanomas associated with nevi. Arch. Dermatol. 2003, 139, 1620–1624; discussion 1624.
- Dessinioti, C.; Antoniou, C.; Katsambas, A.; Stratigos, A.J. Melanocortin 1 receptor variants: Functional role and pigmentary associations. Photochem. Photobiol. 2011, 87, 978–987.
- Rossi, M.; Pellegrini, C.; Cardelli, L.; Ciciarelli, V.; Di Nardo, L.; Fargnoli, M.C. Familial Melanoma: Diagnostic and Management Implications. Dermatol. Pract. Concept 2019, 9, 10–16.
- Goldstein, A.M.; Tucker, M.A. Genetic epidemiology of cutaneous melanoma: A global perspective. Arch. Dermatol. 2001, 137, 1493–1496.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112.
- Eliceiri, B.P.; Klemke, R.; Strömblad, S.; Cheresh, D.A. Integrin alphavbeta3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell Biol. 1998, 140, 1255–1263.
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954.
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350.
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696.
- Scheid, M.P.; Woodgett, J.R. PKB/AKT: Functional insights from genetic models. Nat. Rev. Mol. Cell Biol. 2001, 2, 760–768.
- Stahl, J.M.; Cheung, M.; Sharma, A.; Trivedi, N.R.; Shanmugam, S.; Robertson, G.P. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003, 63, 2881–2890.
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682.
- Kong, Y.; Si, L.; Li, Y.; Wu, X.; Xu, X.; Dai, J.; Tang, H.; Ma, M.; Chi, Z.; Sheng, X.; et al. Analysis of mTOR Gene Aberrations in Melanoma Patients and Evaluation of Their Sensitivity to PI3K-AKT-mTOR Pathway Inhibitors. Clin. Cancer Res. 2016, 22, 1018–1027.
- Goel, V.K.; Lazar, A.J.F.; Warneke, C.L.; Redston, M.S.; Haluska, F.G. Examination of Mutations in BRAF, NRAS, and PTEN in Primary Cutaneous Melanoma. J. Invest. Dermatol. 2006, 126, 154–160.
- Brown, V.L.; Harwood, C.A.; Crook, T.; Cronin, J.G.; Kelsell, D.P.; Proby, C.M. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J. Invest. Dermatol 2004, 122, 1284–1292.
- Alos, L.; Fuster, C.; Castillo, P.; Jares, P.; Garcia-Herrera, A.; Marginet, M.; Agreda, F.; Arance, A.; Gonzalvo, E.; Garcia, M.; et al. TP53 mutation and tumoral PD-L1 expression are associated with depth of invasion in desmoplastic melanomas. Ann. Transl. Med. 2020, 8, 1218.
- Sini, M.C.; Manca, A.; Cossu, A.; Budroni, M.; Botti, G.; Ascierto, P.A.; Cremona, F.; Muggiano, A.; D’Atri, S.; Casula, M.; et al. Molecular alterations at chromosome 9p21 in melanocytic naevi and melanoma. Br. J. Dermatol. 2008, 158, 243–250.
- Fargnoli, M.C.; Gandini, S.; Peris, K.; Maisonneuve, P.; Raimondi, S. MC1R variants increase melanoma risk in families with CDKN2A mutations: A meta-analysis. Eur. J. Cancer 2010, 46, 1413–1420.
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147.
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122.
- Carreira, S.; Goodall, J.; Aksan, I.; La Rocca, S.A.; Galibert, M.D.; Denat, L.; Larue, L.; Goding, C.R. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature 2005, 433, 764–769.
- Piepkorn, M.W.; Longton, G.M.; Reisch, L.M.; Elder, D.E.; Pepe, M.S.; Kerr, K.F.; Tosteson, A.N.A.; Nelson, H.D.; Knezevich, S.; Radick, A.; et al. Assessment of Second-Opinion Strategies for Diagnoses of Cutaneous Melanocytic Lesions. JAMA Netw Open 2019, 2, e1912597.
- Ferrara, G.; Argenziano, G. The WHO 2018 Classification of Cutaneous Melanocytic Neoplasms: Suggestions From Routine Practice. Front. Oncol 2021, 11, 675296.
- WHO. WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; International Agency for Research on Cancer: Lyon, France, 2018; p. 470.
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180.

