Melanoma develops as a result of several genetic alterations, with UV radiation often acting as a mutagenic risk factor. Deep knowledge of the molecular signaling pathways of different types of melanoma allows better characterization and provides tools for the development of therapies based on the intervention of signals promoted by these cascades. The latest World Health Organization classification acknowledged the specific genetic drivers leading to melanoma and classifies melanocytic lesions into nine distinct categories according to the associate cumulative sun damage (CSD), which correlates with the molecular alterations of tumors. The largest groups are melanomas associated with low-CSD or superficial spreading melanomas, characterized by frequent presentation of the BRAFV600 mutation. High-CSD melanomas include lentigo maligna type and desmoplastic melanomas, which often have a high mutation burden and can harbor NRAS, BRAFnon-V600E, or NF1 mutations. Non-CSD-associated melanomas encompass acral and mucosal melanomas that usually do not show BRAF, NRAS, or NF1 mutations (triple wild-type), but in a subset may have KIT or SF3B1 mutations. To improve survival, these driver alterations can be treated with targeted therapy achieving significant antitumor activity. In recent years, relevant improvement in the prognosis and survival of patients with melanoma has been achieved, since the introduction of BRAF/MEK tyrosine kinase inhibitors and immune checkpoint inhibitors.
- melanoma
- molecular pathways
- markers
- target therapy
- BRAF
- NRAS
- KIT
- NTRK
1. Introduction
1.1. Epidemiology
1.2. Risk Factors
2. Molecular Pathways of Melanoma Development
Cancer results from uncontrolled cellular growth of malignant tumor cells caused by a combination of genetic alterations that lead to neoplastic transformation and escape from the inhibitory signals. Several steps in this process are known as the hallmark of cancers [17]. Several key molecular pathways have been discovered to be involved in the onset, proliferation, survival, progression, and invasion. In this section, wresearchers summarize the major signaling pathways that are currently known to be dysregulated and involved in melanoma disease.2.1. MAPK Pathway
Melanomagenesis occurs after mutational events that produce signaling pathways critical for cell survival. Mitogen-activated protein kinase (MAPK) is a signal transduction pathway, involved in a variety of physiological programs, such as cell proliferation, differentiation, development, migration, apoptosis, and transformation, and is the most relevant in the development of melanoma (Figure 1) [18]. The MAPK pathway is activated by the binding of a growth factor to a receptor tyrosine kinase (RTK) on the cell surface and stimulates the guanosine triphosphatases (GTPase) activity of RAS. The signal propagates through the RAF, mitogen-activated protein kinase kinase 1 (MAP2K1), and extracellular signal-related kinase (ERK) cascade, which enters the nucleus to activate transcription factors and promote the cell cycle (Figure 1) [18].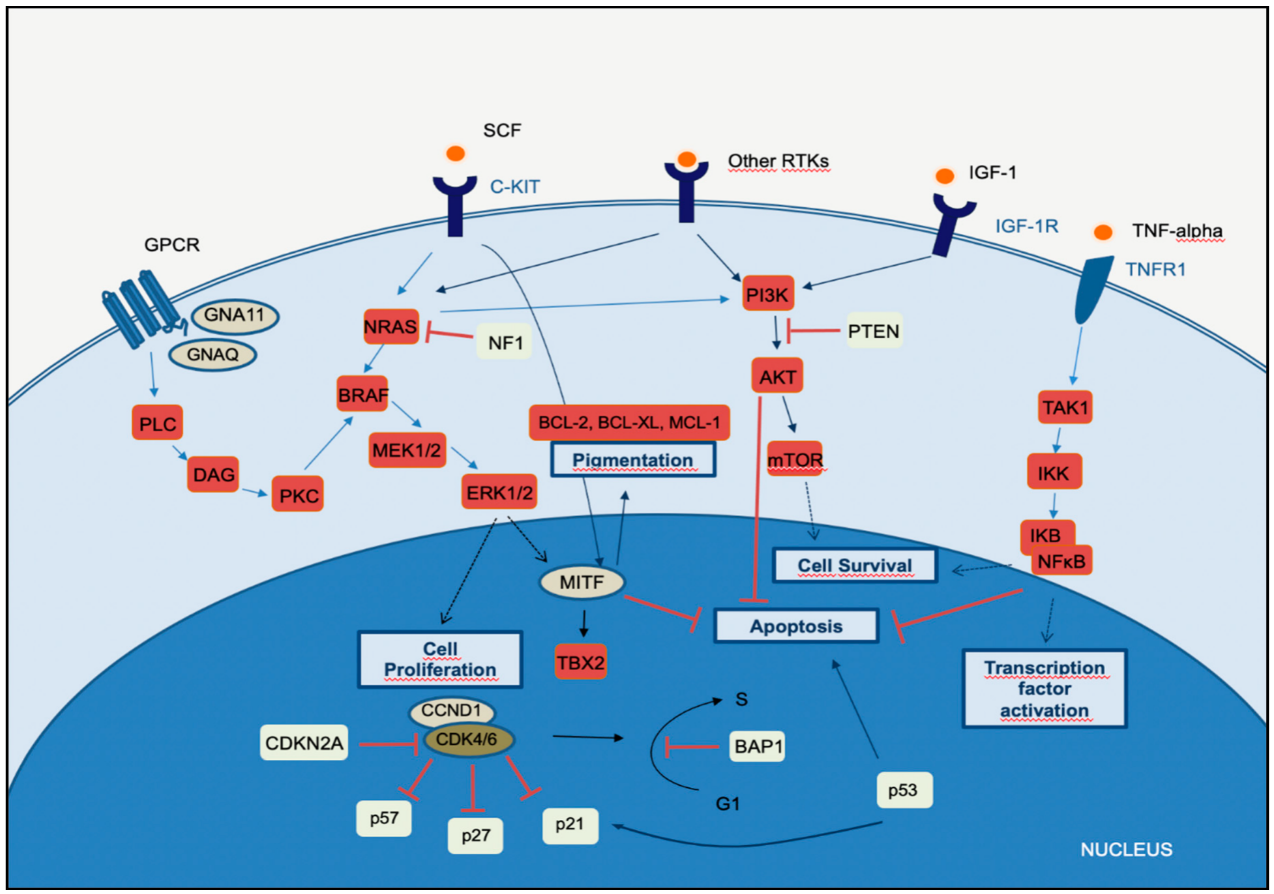
2.2. PI3K-AKT Pathway
The phosphatidylinositol-3-kinases (PI3Ks) comprise a family of lipid kinases with regulatory roles in many cellular mechanisms, including cell survival and growth, differentiation, proliferation, transcription, and translation. The pathway transduces signals from a variety of growth factors and cytokines and is the major downstream effector of RTKs and G-protein-coupled receptors (GPCRs) (Figure 1). Activated PI3K leads to the formation of phosphatidylinositol-3,4,5-trisphosphate (PIP3) through phosphorylation of phosphatidylinositol-4,5-diphosphate (PIP2) in the plasma membrane. PIP3 is essential for the recruitment of the serine-threonine protein kinase AKT to the plasma membrane. AKT is crucial in this signaling pathway, transmitting signals by phosphorylating different downstream effector targets [23]. Once AKT is phosphorylated and fully activated, it turns on a major downstream effector of the PI3K pathway, inhibiting or activating a variety of targets and regulating important cellular processes, such as apoptosis, DNA repair, cell cycle, glucose metabolism, cell growth, motility, invasion, and angiogenesis. The main target of AKT is the mammalian target of rapamycin (mTOR), which has a central role in the PI3K-AKT pathway and cancer disease. mTOR plays a crucial part in regulating cell growth and proliferation by monitoring nutrient availability, cellular energy, oxygen levels, and mitogenic signals. PI3K-AKT signaling has negative regulators, to control any persistent and long-term activation. A major regulator of PI3K-AKT signaling is the tumor suppressor phosphatase and tensin homolog (PTEN), which antagonizes the PI3K activity through its intrinsic lipid phosphatase activity, converting PIP3 back to PIP2. Loss of PTEN results in constitutive activation of AKT and has been largely associated with tumor development in malignant melanoma. Indeed, PTEN loss has been shown to be predictive of shorter overall survival (OS) [24,25][24][25]. The PI3K signaling cascade is upregulated in different types of cancer, including melanoma. More than two-thirds of primary and metastatic melanomas show high levels of phosphorylated AKT, suggesting that this alteration is an early event in melanoma pathogenesis. Oncogenic events that activate PI3K-AKT may include mutations or copy number variations in certain components of the pathway. RAS gene mutations and mutated or amplified expression of RTK may also hyperactivate the PI3K-AKT pathway [20]. Mutations in the mTOR gene are present in approximately 10% of melanomas, and this molecular event leads to shorter survival and worse prognosis [26]. PI3K-AKT signaling may also be activated in melanoma due to loss of function of the negative regulator PTEN, which occurs in 10–30% of cutaneous melanomas, leading to constitutive activation of the PI3K pathway. Interestingly, PTEN gene alterations are mutually exclusive with NRAS mutations, and approximately 20% of melanomas with loss of PTEN function also have BRAFV600E mutations [27].2.3. CDKN2A, Cell Cycle, and Apoptosis Regulation
The CDKN2A gene encodes two proteins, p16CDKN2A and p14CDKN2A, which have a tumor suppressor function. The cyclin proteins bind and activate CDKs, which has catalytic kinase activity. Several cyclin/CDK complexes have been identified that functionally act in different cell cycle phases: in the pre-replicative stage (G1), DNA duplication (S), and promotion of progression through the S phase to mitosis (Figure 1) [28]. p16CDKN2A and p14CDKN2A proteins have an inhibitory function, interfering with the activity of the cyclin/CDK complexes. p16CDKN2A inhibits the cyclin D1 (CCND1)/CDK4 complex, which, in turn, phosphorylates pRb and allows progression through the G1–S checkpoint. p14CDKN2A is an antagonist of the mouse double minute 2 homolog (MDM2) protein. This protein degrades p53 and eliminates p53 control of cell growth. The p14CDKN2A protein inhibits the oncogenic actions of MDM2 by blocking its actions on p53 [28]. p53 is a transcription factor that functions as a major negative regulator of cell proliferation and survival. Inactivation of the TP53 gene results in intracellular accumulation of genetic damage, which promotes melanoma development and progression. TP53 can be inactivated through silencing or mutation, the latter occurring most frequently in high-cumulative solar damage-associated (CSD-associated) melanomas [29]. Somatic impairment of the CDKN2A gene in melanoma can occur by genetic deletions, inactivated mutations, or promoter hypermethylation and leads to a decrease of the function of p16CDKN2A and/or p14CDKN2A proteins, with consequent loss of cell cycle control. This situation is associated with a higher melanoma invasion potential and metastases [30]. As mentioned above, mutation of the CDKN2A gene at the germline level is the most frequent genetic alteration in patients with a strong familial history of melanoma. In addition, variants of the MC1R gene increase the melanoma risk in CDKN2A mutation carriers [31]. The CCND1 and CDK4 genes are found to be altered in a minority of melanomas, representing less than 5%, and depend on the melanoma type. CCND1 gene amplifications affect about 30% of acral melanomas, 11% of lentigo maligna melanomas, and 6% of superficial spreading melanomas. CDK4 gene amplification is frequently found in acral and mucosal melanomas [32].2.4. MITF Pathway
The microphthalmia-associated transcription factor (MITF) acts as a master regulator of melanocyte development, function, and survival by modulating differentiation and cell cycle progression genes [33]. It is involved in the differentiation and maintenance of melanocytes and modulates melanocyte differentiation and pigmentation (Figure 1). In melanomas, MITF can behave as an oncogene, and in approximately 20% of melanomas, it amplifies and promotes the proliferation of tumor cells. Its amplification correlates with a worse prognosis and a lower OS and ChT resistance [33]. MITF is activated by the MAPK and cAMP pathways and regulates the transcription of three major pigmentation enzymes (TYR, TYRP1, and DCT) [34]. In melanoma, ERK activity stimulated by BRAF is associated with MITF ubiquitin-dependent degradation. BRAF can modulate intracellular MITF protein through two opposite mechanisms. On the one hand, it can degrade the MITF protein; on the other hand, BRAF can stimulate transcription factors that increase the expression of the MITF protein. About 10–15% of melanomas harbor the BRAF mutation along with MITF amplification, suggesting that additional mechanisms are involved in ERK-dependent degradation of MITF.3. The Integration of Histology and Molecular Diagnostics of Melanoma
Despite recent molecular advances in melanoma characterization, paramount to diagnosis of a melanocytic skin lesion is the integration of several histopathological criteria with the clinical features. In many cases, general morphological criteria for atypia are often the subject of disagreement and inter-observer variability, especially in non-conventional lesions [41][35]. The World Health Organization (WHO) recognizes these challenges and incorporate the known molecular pathways in the latest WHO melanocytic tumor classification, introducing the concept of “intermediate” lesions. As stated in a recent review on the topic, this multidimensional classification showed that the view of melanocytic tumors as either benign or malignant might no longer be the proper approach [42][36]. Thus, WHO 2018 indicates nine categories/pathways leading to melanoma, each with specific genetic drivers (Table 1). Furthermore, melanomas can be clustered in three main subtypes, according to the degree of CSD (Table 1 and Figure 2) [43][37]. The largest group are melanomas associated with low-CSD or superficial spreading melanomas, which often arise on the trunk and proximal areas of the extremities. The most frequent molecular alteration in these melanomas is the BRAFV600E mutation [44][38]. In addition, TERT promoter mutations and CDKN2A mutations are also found in the majority of cases. PTEN and TP53 are commonly observed in advanced tumors. Lentigo maligna and desmoplastic melanomas are considered tumors associated with high-CSD. These melanomas arise on heavily sun-damaged skin, such as the face or hands, and affect older people. Molecularly, they often have a high mutation load and may harbor NRAS, BRAF non-V600E, or NF1 mutations. TERT promoter mutations and CDKN2A are also frequently found in these melanomas, and KIT mutations are found in a subset of cases. Interestingly, the number of mutations increases with the CSD grade (Figure 2), and desmoplastic melanomas harbor the highest tumor mutation burden. The category of “low to non-UV exposure/CSD” melanomas includes Spitz melanomas, acral melanomas, mucosal melanomas, melanomas developed from congenital nevi and blue nevi, and uveal melanomas. These melanomas rarely harbor BRAF, NRAS, or NF1 mutations (triple wild-type) [43][37]. A subset of acral and mucosal melanomas may have KIT mutations, in addition to gene amplifications and structural rearrangements, most frequently of the CCND1 gene and SF3B1. Therefore, genomic studies have subsequently exemplified that acral and mucosal melanomas are biologically distinct from their cutaneous counterparts at sun-exposed sites. Spitz melanomas show a particular oncogenic signaling pathway involving tyrosine kinase or serine-threonine kinase fusions, and melanomas in blue nevus and uveal melanomas are characterized by GNA11 or GNAQ mutations [44][38].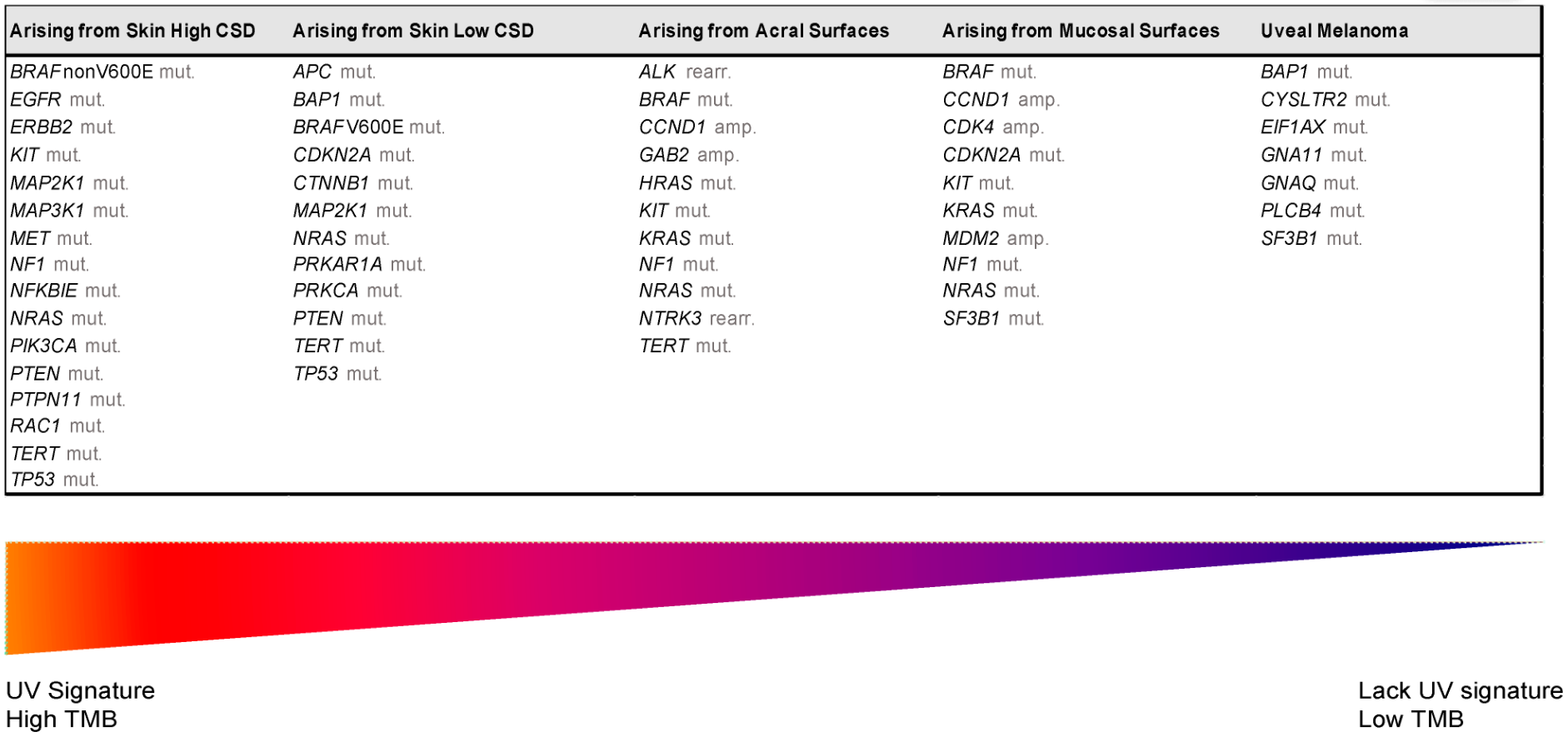
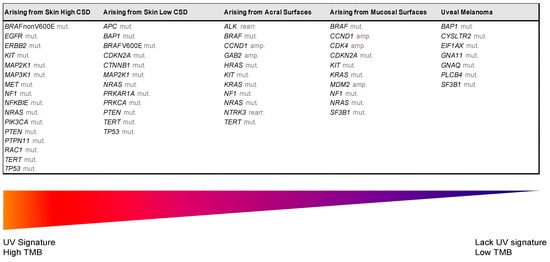
| UV Exposure | Categories | Melanoma Subtype | Key Molecular Genes | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low UV/CSD | I | Superficial spreading melanoma | BRAF | V600 mut | CDKN2A | mut | NRAS | mut | TERT | mut | PTEN | mut | TP53 | mut | ||
| High UV/CSD | II | Lentigo maligna melanoma | NRAS | mut | BRAFnon-V600E | mut | KIT | mut | TERT | mut | CDKN2A | mut | PTEN | mut | TP53 | mut |
| III | Desmoplastic melanoma | NF1 | mut | NFKBIE | mut | NRAS | mut | PIK3CA | mut | |||||||
| Low or no UV/CSD | IV | Spitz melanoma | ALK | rearr | NTRK1 | rearr | NRTK3 | rearr | CDKN2A | mut | HRAS | mut | ||||
| V | Acral melanoma | KIT | mut | NRAS or BRAF | mut | ALK | rearr | NRTK3 | rearr | CDKN2A | mut | CCND1 | amp | TERT | mut | |
| VI | Mucosal melanoma | KIT | mut | NRAS or BRAF | mut | CDKN2A | mut | SF3B1 | mut | CCND1 | amp | CDK4 | mut | MDM2 | amp | |
| VII | Melanoma in congenital nevus | NRAS | mut | BRAFV600E | mut | |||||||||||
| VIII | Melanoma in blue nevus | GNA11 | mut | GNAQ | mut | CYSLTR2 | mut | BAP1 | mut | EIFAX | mut | SF3B1 | mut | |||
| IX | Uveal melanoma | GNA11 | mut | GNAQ | mut | CYSLTR2 | mut | PLCB4 | mut | BAP1 | mut | EIFAX | mut | SF3B1 | mut | |