1. Introduction
The critical intra-cellular signalling dysregulation caused by SARS-CoV-2, which triggers the chronic inflammation that deteriorates into COVID-19
[1][2]. Two effective therapeutic solutions to COVID-19, 28-amino-acid Vasoactive Intestinal Peptide and 14-amino-acid Human Ezrin Peptides, seem to have related mechanisms of action, which may be based on TIRAP inhibition, PKA activation, and increased transcription-factor CREB-mediated expression. These therapeutic peptides not only stop the virus-triggered innate-immune-activation, which is the cause of the destructive COVID-19 inflammatory-disease progression, they also amplify adaptive anti-viral B-Cell and T-Cell immune responses that inhibit the SARS-CoV-2 virus life cycle and prevent re-infection.
Regulated innate and adaptive immune responses are essential to limit viral infections and stop inflammation-related tissue damage. Innate immunity is the ancient first line of defence, which is mobilised within minutes against viral, bacterial, or fungal invasion of multicellular organisms. The innate immune system evolved with multicellular animal species to defend against invading microorganisms, mainly by detecting characteristic conserved carbohydrate and lipid polymers that display Pathogen-Associated-Molecular-Patterns (PAMPs), which are recognised by genetically pre-programmed Pattern Recognition Receptors (PRRs), for example the Toll-Like-Receptors (TLRs).
2. Ezrin Inflammation Control
2.1. Introduction to Ezrin
The structures and functions of multi-protein-complexes built by ezrin at the cell membrane are generally over-simplified in the literature, but the data suggests co-ordination of multiple conformations of proteins and multiple phosphorylations of specific residues.
The evolution-conserved Ezrin, Radixin, and Moesin (ERM) adaptor-proteins are membrane-associated multi-protein-complex-formers, receptor and signalling proteins, which also regulate and anchor filamentous F-actin to the cell membrane. Ezrin has a key role in the regulation of alveolar structure and lung homeostasis.
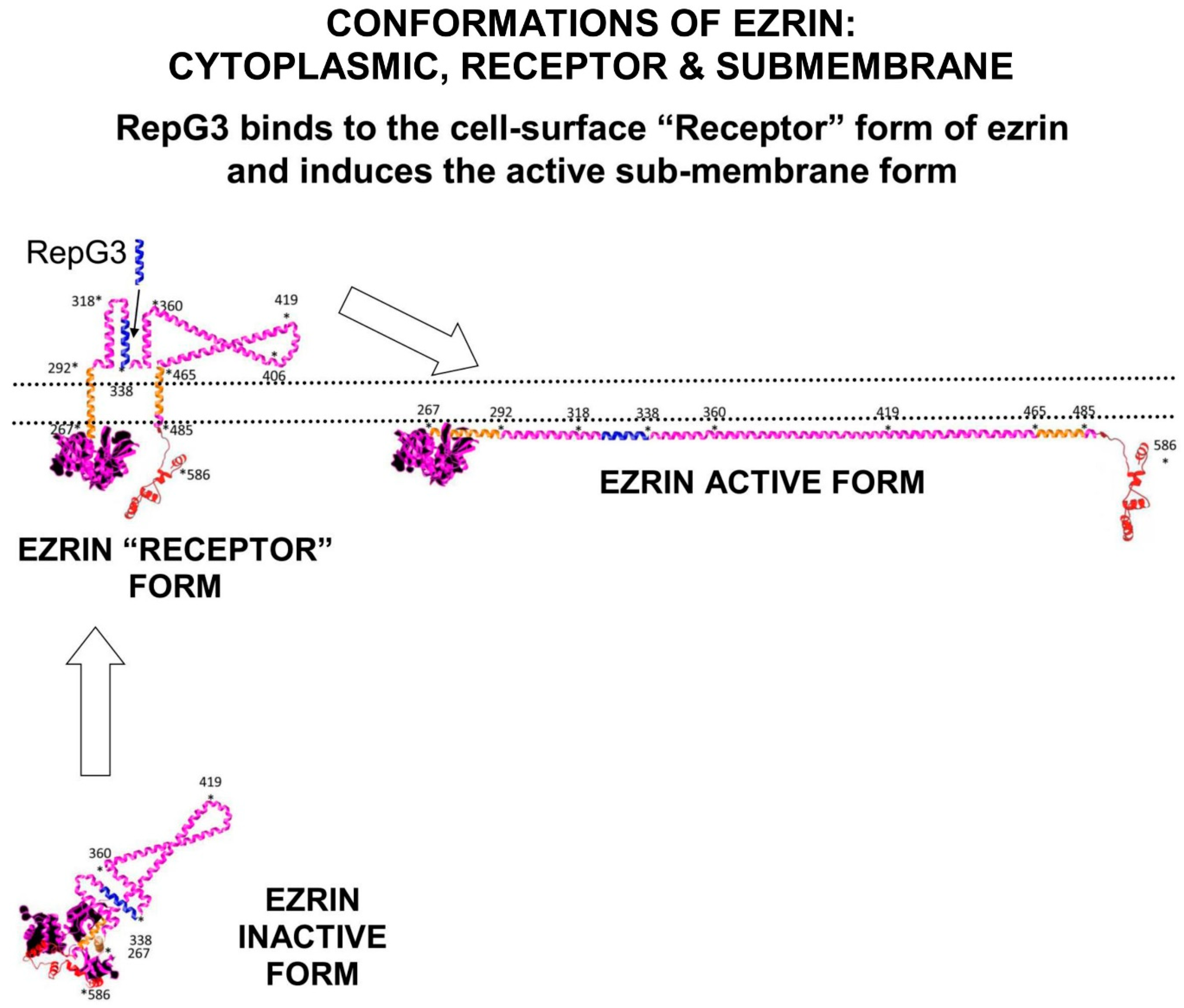
Ezrin is a sub-membrane adaptor protein for multi-protein cell signalling complexes and depending on its conformation can also function as a cell-surface-receptor. Ezrin consists of three domains: The N-terminal FERM-domain formed into a clover-leaf-like structure with three lobes (F1 lobe; amino-acids 1–93: F2 lobe; amino-acids 94–200: F3 Lobe amino-acids 201–296). The FERM-domain is connected to a folded α-helical Alpha domain (ALPHA; amino-acids 297–468), a transmembrane proline-rich domain (TRANS amino-acids 469–497), and a C-terminal domain (C-terminal amino-acids 498–585)
[3].
Activation of ezrin results in a shift of ezrin from the cytoplasm to membrane microvilli, ruffles, pits, and adhesion sites. In the dormant soluble cytoplasmic form of ezrin, the C-terminal domain is bound to the FERM domain between the F2 and F3 lobes. The first stage of ezrin activation occurs at the sub-surface of the cell membrane, where the F1 and F3 lobes of the FERM domain binds to sub-membrane surface PIP2-lipid-rafts and partially unfolds.
Ezrin probably inserts the folded Alpha domain through the membrane at this step, creating the “Receptor” conformation of ezrin. Ezrin adopts its fully elongated conformation after kinase phosphorylation of Thr567 on the C-terminal domain. Depending on the signalling environment, this phosphorylation is performed by either ROCK, or PKCα, or Akt2 or GRK2 (Figure 1).
In its fully unfolded “Active” conformation, ezrin stretches out under the sub-membrane surface and exposes multiple binding sites on its FERM-domain. Elongated ezrin can form multi-protein complexes with a range of receptors, adhesion proteins, ion channel proteins, and cell signalling kinases. Ezrin has specific binding sites on its FERM-domain for sub-membrane-PIP2 lipid-rafts, and the cytoplasmic tails of adhesion molecules and growth-factor receptors such as CD44, CD43, ICAM1&2, and EGFR.
In addition, there are also specific binding sites on the ezrin-FERM domain for cell-signalling proteins such as SOS and Ras. The FERM-domain also binds to NHERF, which binds the ion channel proteins NHE and CFTR. On the ezrin-Alpha domain there are binding sites for the cell signalling proteins PKC, PI3K, and PKA. On the c-terminal domain, there are binding sites for F-actin
[4].
Ezrin is inactive in the cytoplasm, but upon phosphorylation on serine and threonine residues by PKC or RhoA kinase, the protein translocates to the cell surface where it builds multi-protein-complexes that connect to the actin cytoskeleton.
Figure 1. Ezrin has three main conformations: a soluble globular inactive form in the cytoplasm; a membrane associated “Receptor” form at the membrane where the Alpha domain is inserted through the membrane; and an elongated activated form which is attached to PIP2-lipid rafts on the submembrane and stabilised by T567 phosphorylation. The “Receptor” transition conformation of ezrin with part of the Alpha domain exposed on the cell surface can be detected by anti-ezrin alpha-domain monoclonal antibody [6F1A9] (ab205381)
[5]. RepG3, a peptide derived from the ezrin-Alpha domain, triggers a change in conformation from the “Receptor” form to the sub-membrane active form of ezrin. The main phosphorylation sites on ezrin are: tyrosine Y146, tyrosine Y354, Serine S366, tyrosine Y478, Serine S535, and Threonine T567 (phosphorylated by ROCK2 and PKC/PRKCI)
[6]. “*” means N-to-C amino acid sequence number.
2.2. Ezrin Regulates m-RAGE+TIRAP Mediated Inflammation
m-RAGE, CD44, and ezrin are important regulators that maintain normal alveolar epithelial structure and function in the lung. In resting alveolar-epithelial A549 cells, m-RAGE and ezrin are abundantly expressed together in the cell-surface microvilli and also co-localise as protein–protein complexes diffusely distributed in the cytoplasm. In these m-RAGE+ezrin complexes, the FERM domain and the C-terminal domain of ezrin both bind to the cytoplasmic tail of m-RAGE (ezrin is in its transition “Receptor” conformation). Pro-inflammatory cytokine treatment of A549 cells (for example a mixture of TGF-1, IL-6, IL-1, and TNFa) causes more ezrin to relocate from the cytoplasm to the sub-surface of the cell membrane, and triggers ROCK to phosphorylate C-terminal T567 of ezrin
[7]. This threonine-phosphorylation switches the conformation of ezrin from its “Receptor” conformation to stretch-out into its sub-membrane “Active” conformation, which disrupts ezrin+m-RAGE complexes clustered on the sub-membrane. The m-RAGE+ezrin complexes also disappear from the cytoplasm.
In addition, TLR4 mediated LPS-induced p38MAPK activation and nuclear NFκB activation is ezrin dependent. Ezrin associates with MyD88/IRAK-1 upon LPS challenge. Blockade of RhoA/ROCK inhibits LPS-induced ezrin phosphorylation and its translocation from the cytoplasm to the cell membrane. Moreover, the suppression of ezrin by siRNA or the blockade of ROCK activation with Y-27632 reduces the production of TNF-α, IL-1β, and HMGB1 in response to LPS
[8].
High concentrations of negatively charged PIP2 on the sub-surface of the plasma-membrane are required for multi-protein-complex formation. The local sub-membrane concentrations of PIP2 controls the activation state of both TLR4+TIRAP+MyD88 and m-RAGE+TIRAP+MyD88 multi-protein-complexes. Ezrin also occupies PIP2-lipid-rafts to anchor its own multi-protein-complexes.
Lipopolysaccharide (LPS) triggering of TLR4 leads to the formation of the TLR4+TIRAP+MyD88 complex. The association of TIRAP and MyD88 into a multi-protein-complex depends on the availability of lipid-rafts containing high concentrations of PIP2. A critical step is the electrostatic attraction of negatively charged PIP2 in the sub-membrane, for positively charged TIRAP. The N-terminal region of TIRAP contains a PIP2-binding domain (PBD) that has a strong binding affinity for PIP2, which induces conformational changes in TIRAP that results in its activation.
PIP2 is synthesized by membrane-associated PIP5Kα, which is an essential component of TIRAP-dependent TLR4-activation. LPS triggering of TLR4 increases PIP5Kα mediated synthesis of PIP2. Higher PIP2 concentrations in the lipid-rafts induces more TIRAP translocation to the plasma membrane. PIP5Kα knock-down results in lower PIP2 concentrations in the sub-membrane and significantly attenuates TLR4-induced phosphorylation of p38MAPK, JNK, and NFκB, which inhibits the expression of pro-inflammatory cytokines IL-6 and TNF-α. In addition, Phospholipase-γ2 (PLCγ2) decreases the lipid-raft concentration of PIP2. Low concentrations of PIP2 results in TLR4+TIRAP+MyD88 complexes being internalized by endocytosis and their dissociation into separate MyD88, TIRAP, and TLR4 proteins
[9].
TIRAP specifically localizes to the zones of the cell membrane where activated-ezrin binds PIP2. High concentrations of PIP2 in lipid rafts are also necessary for ezrin multi-protein-complex formation that are involved in actin-cytoskeleton re-arrangement and the anchoring of filamentous-actin at “membrane ruffles”.
The FERM-domain of activated-ezrin is a competitive-inhibitor of TIRAP and activated ezrin regulates the activity of the multi-protein-complexes of m-RAGE and TLR4+TIRAP+MyD88 by competing with TIRAP for space on the sub-membrane lipid-rafts of concentrated PIP2. This competition for PIP2-lipid-rafts between TIRAP and ezrin is a “protein-complex-switch” for turning inflammatory signalling on-and-off. Ezrin has also been demonstrated to regulate inflammatory cytokine expression in B-cells. Conditional deletion or chemical inhibition of active ezrin in B-cells increases LPS-triggered TLR4-induction of pro-inflammatory cytokine production
[10].
2.3. Ezrin+S100 Complexes Regulate S100+RAGE Inflammation
When S100 proteins bind to m-RAGE, they activate inflammation. When S100 proteins bind to ezrin, inflammatory signalling is suppressed
[11]. However, ezrin binds to S100-proteins involved in m-RAGE activation in an unexpected way. In HEK-293T cells, S100A4 co-localizes below the cell membrane in regions rich in ezrin. However, NMR spectroscopy in vitro of S100A4+ezrin interactions revealed that S100 proteins efficiently bind to both the F2-lobe of the isolated N-terminal FERM domains of ezrin and also to isolated small C-terminal actin-binding domains of ezrin, with similar high-affinity. However, S100A4 only very weakly binds to sub-membrane associated full-length ezrin, nor does it bind to the fully folded inactive soluble cytoplasmic conformation of ezrin either.
Fast kinetic measurements reveal that S100A4 binding induces structural changes to the whole of the ezrin protein. The physiological complex of S100+ezrin probably involves the ezrin “Receptor” conformation, where the FERM domain and C-terminal domain are loosely associated below the cell membrane and the ezrin-Alpha domain is exposed on the cell surface.
In the cytoplasm of many cell types, there are inactive complexes of ezrin+S100-Calgranulin-C (S100A12), where ezrin is in the “Receptor” transition-state between conformations. When intracellular calcium concentration increases, S100A12+ezrin binds Ca2+ in the cytoplasm and then translocates to the cell-membrane. Calcium-bound S100A12 forms dimeric or even hexameric structures on the sub-membrane surface. In the presence of ezrin, m-RAGE is also internalized into the cell and then recycled back to the cell membrane, after binding to S100+ezrin complexes.
2.4. The Ezrin-Protein-Complex and the Ras>Raf>MEK>ERK>RSK Pathway
Activated phospho-ezrin+actin catalyses the formation of a multi-protein-complex consisting of Receptor Tyrosine Kinases (RTKs) with Grb2, SOS, and Ras. The small G-protein p21-Ras binds to the ezrin FERM domain and other signalling proteins, where it functions as a molecular-switch, relaying extracellular stimuli to diverse intracellular effector pathways, which are responsible for controlling proliferation, motility, and differentiation
[12].
SOS catalyses the nucleotide-exchange on Ras but the activation of Ras also requires the participation of ezrin and co-receptors such as CD44 and actin. Disrupting the interaction between ezrin and co-receptors, for example by down-regulation of different ERM proteins by siRNA, or by expression of dominant-negative mutants, or by disruption of F-actin, abolishes growth-factor-induced Ras-activation. In addition, over-expression of ezrin-R579A mutant, that is unable to bind F-actin, also reduces RTK-Ras dependent signalling, and IL-6-dependent ERK phosphorylation. In contrast, ezrin-R579A does not interfere with Ezrin-PKC-induced ERK phosphorylation.
Triggering of Grd2+SOS+ezrin+CD44+actin+p21Ras>Raf>MEK>ERK>p90RSK signalling results in the activation of transcription-factor CREB, inhibition of NFκB, and inhibition of pro-inflammatory cytokine expression.
3. VASO-Active Intestinal Peptide and COVID-19
3.1. Natural Plasma-VIP Levels in Patients Correlate to Recovery from Severe COVID-19
The plasma concentration of natural-VIP is elevated in the plasma of individuals with manifestations of severe COVID-19, and the amount of VIP-elevation correlates to survival and recovery from severe COVID-19
[13].
In a clinical study in April and May 2020, with 24 critically ill COVID-19 patients of median age 53 years, the plasma levels of VIP were measured and compared with patients with only mild COVID-19 symptoms, and with non-infected healthy individuals. As expected, comparing five pro-inflammatory markers (TNF-α IL-8, IL-12p40, IL-17A, CXCL10/IP-10) in mild COVID-19 and severe COVID-19 cases, there was a significant positive correlation between pro-inflammatory cytokine concentration and severity of disease symptoms. Severe COVID-19 was characterized by very elevated serum levels of pro-inflammatory cytokines, particularly IL-6, TNFa, IL-8, and IL-1β.
Plasma levels of VIP were significantly elevated in patients with severe COVID-19. However, in the group of severe COVID-19 patients, higher levels of VIP correlated with lower levels of pro-inflammatory cytokines. The critical discovery was that high VIP plasma levels significantly correlated with survival of patients from severe COVID-19, compared to patients who died of severe COVID-19.
3.2. VIP In Vitro Inhibits SARS-CoV-2-Induced NFκB Activation and IL-6 Expression
In vitro in infected monocytes, SARS-CoV-2 activates NFκB to increase the amounts of the pro-inflammatory cytokines above controls; IL-6 up 15x, IL-8 up 4x, and TNFa up 12x. Phosphorylated NFκBp65 subunit, a marker for activated NFκB, was also significantly up-regulated in SARS-CoV-2 infected cells. In vitro treatment with VIP reduced the NFκB p65 phosphorylation.
Calu-3 cells, a lung epithelial cell line which expresses VIP-receptor VPAC1, are highly susceptible to SARS-CoV-2 infection. In Calu-3 lung epithelial cells, VIP inhibited activation of NFκB and reduced pro-inflammatory cytokine expression by about fifty per cent, which correlated with reduced viral production and protection from cell death. A specific inhibitor of NFκB was found to mimic the anti-inflammatory activity of VIP. Treatment of SARS-CoV-2-infected monocytes with Bay11-7082, a pharmacological inhibitor of NFκB, resulted in decreased viral RNA synthesis, reduced cell death, and suppression of production of IL-6 and TNF-α.
3.3. VIP In Vitro Decreases SARS-CoV-2 Replication
In vitro, VIP decreases SARS-CoV-2 replication and viral mRNA expression in human monocytes and Calu-3 lung epithelial cells. Treatment of SARS-CoV-2 infected Calu-3 cells with 10 nM VIP results in a fifty per cent reduction in SARS-CoV-2 RNA synthesis. However, it was intriguing that when comparing mild COVID-19 and severe COVID-19 in patients, there was only a weak correlation between plasma VIP levels and SARS-CoV-2 viral load.
3.4. VIP Up-Regulates CREB to Inhibit NFκB Mediated Inflammation
Cyclic-AMP Response Element Binding-protein (CREB) is a phosphorylation-activated transcription-factor, part of the basic-leucine-zipper (bZIP) superfamily, comprising of CREB, ATF1, and CREM. CREB has an alpha-helical structure, which contains a Kinase Inducible Domain (KID) in its central region, two glutamate Q1 and Q2 domains and a bZIP domain. The phosphorylation site located at Serine-133 (Ser133) on CREB is critical for CREB transcriptional activity. CREB can be phosphorylated at Ser133 by the serine-threonine kinases PKA, PKC, and RSK. Activated CREB can form homodimers or heterodimers in a transcription-complex with CREB-Binding-Protein (CBP) and p300 (also called EP300 or E1A binding protein p300), which bind to DNA at Cyclic-AMP Response Element CRE-promotors, initiating transcription of CREB responsive genes.
Effective NFκB transcription of pro-inflammatory cytokine genes requires interaction of the NFκB subunit p65 (RelA) with either CBP or p300. However, phosphorylation of CREB-serine133 in its kinase-inducible-domain (KID) promotes the binding of the co-activator proteins CBP and p300 to activated-CREB to commence transcription of CREB-responsive genes. Activated-CREB inhibits NFκB mediated transcription by sterically hindering the binding of CBP to the NFκB complex. Activated-CREB also starves NFκB of CBP and p300, preventing it functioning as a transcription-factor.
In SARS-CoV-2-infected monocytes, CREB activation and its transcription-factor mediated gene-expression are suppressed by the viral proteins in control of cell-signalling, which chronically amplify NFκB activity. In contrast, treatment of SARS-CoV-2-infected monocytes with VIP triggers the activation of transcription-factor CREB. The activated-CREB in these virally infected cells inhibits the activation of transcription-factor NFκB that is responsible for the expression of pro-inflammatory cytokines. CREB activation is also connected to an anti-apoptotic response in monocytes and macrophages exposed to inflammatory stimuli. In addition, CREB may also directly suppress SARS-CoV-2 viral replication by inhibition of cell signalling pathways that lead to NFκB activation. CREB also promotes anti-inflammatory immune responses, through the induction of anti-inflammatory IL-10.
3.5. Intravenous VIP-Therapy Rescues Patients from Severe-COVID-19
Aviptadil, a synthetic form of Vasoactive Intestinal Peptide (VIP) has been granted Fast Track Designation for the treatment of Critical COVID-19 with Respiratory Failure.
In a controlled clinical trial at Houston Methodist Hospital, intravenous treatment of patients suffering severe COVID-19 with Vasoactive Intestinal Peptide (synthetic-VIP, brand-name Aviptadil) was performed in a prospective, open-label clinical study. The 45 patients enrolled in the study were admitted to the intensive care unit (ICU) in June and July 2020 with severe/critical COVID-19 and co-morbidities
[14][15][16].
The 21 patients in the VIP treatment group were unconscious when treated with Aviptadil, so the chance of placebo bias was discounted because they could not know which medication they were receiving through multiple intravenous lines. Patients in the VIP treatment group received three successive 12-h intravenous infusions of VIP-Aviptadil at 50 pmol/kg/h, 100 pmol/kg/h, and 150 pmol/kg/h. The main clinical outcomes measured included survival and recovery from respiratory failure.
However, Aviptadil has side-effects; it is known to cause serious hypotension and diarrhoea, so intravenous VIP should only be administered by critical-care physicians who have sufficient expertise to manage these side effects. About twenty per cent of patients exhibited VIP-related hypotension and/or diarrhoea, but no other serious VIP-related adverse events were recorded.
Patients treated with intravenous Aviptadil-VIP were followed for at least 60 days after ICU admission. They were compared with 24 individuals with comparable COVID-19 and co-morbidities, who received only standard-of-care treatment. Of the 21 patients treated with Aviptadil, 19 survived to day 28, 17 survived to day 60 compared to only five survivors of the 24 individuals in the standard-therapy group (81% vs. 21%; p < 0.0001). Improved radiographic appearance was seen in both lungs of 17 VIP treated patients but only one lung of two VIP treated patients who subsequently died. Four out of five patients treated with Aviptadil on Extracorporeal Membrane Oxygenation were successfully de-cannulated and survived. In comparison, only three out of 13 standard-therapy patients survived.
Initial release of the data revealed increased survival rates that correlated to a significant reduction in plasma IL-6 and improved lung-surfactant production.
In all patients, the reduction of the inflammatory markers C-reactive protein (−76% ± 3%) and IL-6 (−75% ± 3%) was very significant.
 +1 credit
+1 credit