SARS-CoV-2 infection inhibits interferon expression, while hyper-activating innate-immune signalling and expression of pro-inflammatory cytokines. SARS-CoV-2 proteins: Spike, M and nsp6, nsp12 and nsp13 inhibit IFR3-mediated Type-1-interferon defence, but hyper-activate intracellular signalling, which leads to dysfunctional expression of pro-inflammatory cytokines, particularly IL-1β IL-6, IL-8, and TNFα. Ezrin, a sub-membrane adaptor-protein, organises multi-protein-complexes such as ezrin+NHERF1+NHE+CFTR, which control the density and location of ACE2 receptor expression on the luminal surface of airway-epithelial-cells, as well as determining susceptibility to SARS-CoV-2 infection. This protein complex is vital for lung-surfactant production for efficient gas-exchange. Ezrin also forms multi-protein-complexes that regulate signalling kinases; Ras, PKC, PI3K, and PKA. m-RAGE is a pattern-recognition-receptor of the innate immune system that is triggered by AGEs, which are chemically modified proteins common in the elderly and obese. Vasoactive Intestinal Peptide (VIP) is a 28-amino acid peptide released by intrinsic neurons in the human airways.
- COVID-19
- Ezrin
- Vasoactive Intestinal Peptide
1. Introduction
2. Ezrin Inflammation Control
2.1. Introduction to Ezrin
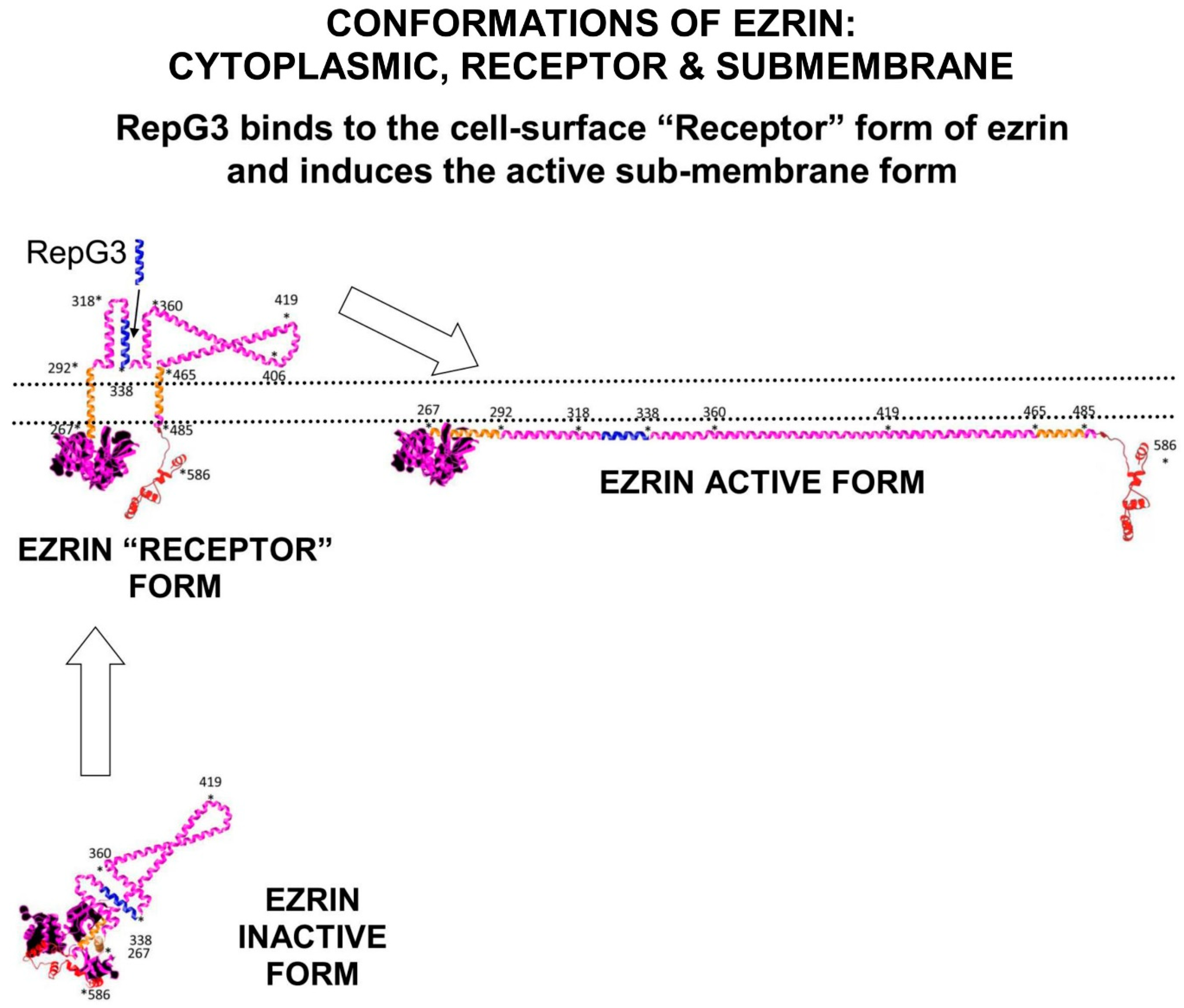
2.2. Ezrin Regulates m-RAGE+TIRAP Mediated Inflammation
2.3. Ezrin+S100 Complexes Regulate S100+RAGE Inflammation
2.4. The Ezrin-Protein-Complex and the Ras>Raf>MEK>ERK>RSK Pathway
3. VASO-Active Intestinal Peptide and COVID-19
3.1. Natural Plasma-VIP Levels in Patients Correlate to Recovery from Severe COVID-19
The plasma concentration of natural-VIP is elevated in the plasma of individuals with manifestations of severe COVID-19, and the amount of VIP-elevation correlates to survival and recovery from severe COVID-19 [13]. In a clinical study in April and May 2020, with 24 critically ill COVID-19 patients of median age 53 years, the plasma levels of VIP were measured and compared with patients with only mild COVID-19 symptoms, and with non-infected healthy individuals. As expected, comparing five pro-inflammatory markers (TNF-α IL-8, IL-12p40, IL-17A, CXCL10/IP-10) in mild COVID-19 and severe COVID-19 cases, there was a significant positive correlation between pro-inflammatory cytokine concentration and severity of disease symptoms. Severe COVID-19 was characterized by very elevated serum levels of pro-inflammatory cytokines, particularly IL-6, TNFa, IL-8, and IL-1β. Plasma levels of VIP were significantly elevated in patients with severe COVID-19. However, in the group of severe COVID-19 patients, higher levels of VIP correlated with lower levels of pro-inflammatory cytokines. The critical discovery was that high VIP plasma levels significantly correlated with survival of patients from severe COVID-19, compared to patients who died of severe COVID-19.3.2. VIP In Vitro Inhibits SARS-CoV-2-Induced NFκB Activation and IL-6 Expression
In vitro in infected monocytes, SARS-CoV-2 activates NFκB to increase the amounts of the pro-inflammatory cytokines above controls; IL-6 up 15x, IL-8 up 4x, and TNFa up 12x. Phosphorylated NFκBp65 subunit, a marker for activated NFκB, was also significantly up-regulated in SARS-CoV-2 infected cells. In vitro treatment with VIP reduced the NFκB p65 phosphorylation. Calu-3 cells, a lung epithelial cell line which expresses VIP-receptor VPAC1, are highly susceptible to SARS-CoV-2 infection. In Calu-3 lung epithelial cells, VIP inhibited activation of NFκB and reduced pro-inflammatory cytokine expression by about fifty per cent, which correlated with reduced viral production and protection from cell death. A specific inhibitor of NFκB was found to mimic the anti-inflammatory activity of VIP. Treatment of SARS-CoV-2-infected monocytes with Bay11-7082, a pharmacological inhibitor of NFκB, resulted in decreased viral RNA synthesis, reduced cell death, and suppression of production of IL-6 and TNF-α.3.3. VIP In Vitro Decreases SARS-CoV-2 Replication
In vitro, VIP decreases SARS-CoV-2 replication and viral mRNA expression in human monocytes and Calu-3 lung epithelial cells. Treatment of SARS-CoV-2 infected Calu-3 cells with 10 nM VIP results in a fifty per cent reduction in SARS-CoV-2 RNA synthesis. However, it was intriguing that when comparing mild COVID-19 and severe COVID-19 in patients, there was only a weak correlation between plasma VIP levels and SARS-CoV-2 viral load.3.4. VIP Up-Regulates CREB to Inhibit NFκB Mediated Inflammation
Cyclic-AMP Response Element Binding-protein (CREB) is a phosphorylation-activated transcription-factor, part of the basic-leucine-zipper (bZIP) superfamily, comprising of CREB, ATF1, and CREM. CREB has an alpha-helical structure, which contains a Kinase Inducible Domain (KID) in its central region, two glutamate Q1 and Q2 domains and a bZIP domain. The phosphorylation site located at Serine-133 (Ser133) on CREB is critical for CREB transcriptional activity. CREB can be phosphorylated at Ser133 by the serine-threonine kinases PKA, PKC, and RSK. Activated CREB can form homodimers or heterodimers in a transcription-complex with CREB-Binding-Protein (CBP) and p300 (also called EP300 or E1A binding protein p300), which bind to DNA at Cyclic-AMP Response Element CRE-promotors, initiating transcription of CREB responsive genes. Effective NFκB transcription of pro-inflammatory cytokine genes requires interaction of the NFκB subunit p65 (RelA) with either CBP or p300. However, phosphorylation of CREB-serine133 in its kinase-inducible-domain (KID) promotes the binding of the co-activator proteins CBP and p300 to activated-CREB to commence transcription of CREB-responsive genes. Activated-CREB inhibits NFκB mediated transcription by sterically hindering the binding of CBP to the NFκB complex. Activated-CREB also starves NFκB of CBP and p300, preventing it functioning as a transcription-factor. In SARS-CoV-2-infected monocytes, CREB activation and its transcription-factor mediated gene-expression are suppressed by the viral proteins in control of cell-signalling, which chronically amplify NFκB activity. In contrast, treatment of SARS-CoV-2-infected monocytes with VIP triggers the activation of transcription-factor CREB. The activated-CREB in these virally infected cells inhibits the activation of transcription-factor NFκB that is responsible for the expression of pro-inflammatory cytokines. CREB activation is also connected to an anti-apoptotic response in monocytes and macrophages exposed to inflammatory stimuli. In addition, CREB may also directly suppress SARS-CoV-2 viral replication by inhibition of cell signalling pathways that lead to NFκB activation. CREB also promotes anti-inflammatory immune responses, through the induction of anti-inflammatory IL-10.3.5. Intravenous VIP-Therapy Rescues Patients from Severe-COVID-19
Aviptadil, a synthetic form of Vasoactive Intestinal Peptide (VIP) has been granted Fast Track Designation for the treatment of Critical COVID-19 with Respiratory Failure. In a controlled clinical trial at Houston Methodist Hospital, intravenous treatment of patients suffering severe COVID-19 with Vasoactive Intestinal Peptide (synthetic-VIP, brand-name Aviptadil) was performed in a prospective, open-label clinical study. The 45 patients enrolled in the study were admitted to the intensive care unit (ICU) in June and July 2020 with severe/critical COVID-19 and co-morbidities [14][15][16]. The 21 patients in the VIP treatment group were unconscious when treated with Aviptadil, so the chance of placebo bias was discounted because they could not know which medication they were receiving through multiple intravenous lines. Patients in the VIP treatment group received three successive 12-h intravenous infusions of VIP-Aviptadil at 50 pmol/kg/h, 100 pmol/kg/h, and 150 pmol/kg/h. The main clinical outcomes measured included survival and recovery from respiratory failure. However, Aviptadil has side-effects; it is known to cause serious hypotension and diarrhoea, so intravenous VIP should only be administered by critical-care physicians who have sufficient expertise to manage these side effects. About twenty per cent of patients exhibited VIP-related hypotension and/or diarrhoea, but no other serious VIP-related adverse events were recorded. Patients treated with intravenous Aviptadil-VIP were followed for at least 60 days after ICU admission. They were compared with 24 individuals with comparable COVID-19 and co-morbidities, who received only standard-of-care treatment. Of the 21 patients treated with Aviptadil, 19 survived to day 28, 17 survived to day 60 compared to only five survivors of the 24 individuals in the standard-therapy group (81% vs. 21%; p < 0.0001). Improved radiographic appearance was seen in both lungs of 17 VIP treated patients but only one lung of two VIP treated patients who subsequently died. Four out of five patients treated with Aviptadil on Extracorporeal Membrane Oxygenation were successfully de-cannulated and survived. In comparison, only three out of 13 standard-therapy patients survived. Initial release of the data revealed increased survival rates that correlated to a significant reduction in plasma IL-6 and improved lung-surfactant production. In all patients, the reduction of the inflammatory markers C-reactive protein (−76% ± 3%) and IL-6 (−75% ± 3%) was very significant.References
- Trougakos, I.P.; Stamatelopoulos, K.; Terpos, E.; Tsitsilonis, O.E.; Aivalioti, E.; Paraskevis, D.; EfsKastritis, E.; Pavlakis, G.N.; Dimopoulos, M.A. Insights to SARS-CoV-2 life cycle, pathophysiology, and rationalized treatments that target COVID-19 clinical complications. J. Biomed. Sci. 2021, 28, 9.
- Choudhary, S.; Sharma, K.; Silakari, O. The interplay between inflammatory pathways and COVID-19: A critical review on pathogenesis and therapeutic options. Microb. Pathog. 2021, 150, 104673.
- Bretscher, A.; Reczek, D.; Berryman, M. Ezrin: A protein requiring conformational activation to link microfilaments to the plasma membrane in the assembly of cell surface structures. J. Cell Sci. 1997, 110, 3011–3018.
- Ivetic, A.; Ridley, A.J. Ezrin/radixin/moesin proteins and Rho GTPase signalling in leucocytes. Immunology 2004, 112, 165–176.
- Company Product Information. Available online: https://www.abcam.com/ezrin-antibody-6f1a9-ab205381.html (accessed on 16 February 2022).
- UniProt Information on Human Ezrin UniProtKB—P15311, (EZRI_HUMAN). Available online: https://www.uniprot.org/uniprot/P15311 (accessed on 16 February 2022).
- Buckley, S.T.; Medina, C.; Kasper, M.; Ehrhardt, C. Interplay between RAGE, CD44, and focal adhesion molecules in epithelial-mesenchymal transition of alveolar epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L548–L559.
- Ding, N.; Li, H.; Gao, B.; Lei, Y.; Zhang, Z. The ROCK-Ezrin Signaling Pathway Mediates LPS Induced Cytokine Production in A549 Cells. Res. Sq. 2021, 1–21.
- Le, O.T.T.; Nguyen, T.T.N.; Lee, S.Y. Phosphoinositide turnover in Toll-like receptor signaling and trafficking. BMB Rep. 2014, 47, 361–368.
- Pore, D.; Matsui, K.; Parameswaran, N.; Gupta, N. Cutting Edge: Ezrin Regulates Inflammation by Limiting B Cell IL-10 Production. J. Immunol. 2015, 196, 558–562.
- Biri-Kovacs, B.; Kiss, B.; Vadászi, H.; Gógl, G.; Pálfy, G.; Török, G.; Homolya, L.; Bodor, A.; Nyitray, L. Ezrin interacts with S100A4 via both its N- and C-terminal domains. PLoS ONE 2017, 12, e0177489.
- Sperka, T.; Geißler, K.J.; Merkel, U.; Scholl, I.; Rubio, I.; Herrlich, P.; Morrison, H.L. Activation of Ras Requires the ERM-Dependent Link of Actin to the Plasma Membrane. PLoS ONE 2011, 6, e27511.
- Temerozo, J.; Sacramento, C.; Fintelman-Rodrigues, N. IP plasma levels associate with survival in 1 severe COVID-19 patients, correlating with protective effects in SARS-CoV-2-infected cells. bioRxiv 2021.
- Youssef, J.; Javitt, J.; Lavin, P.; Al-Saadi, M.; Zahiruddin, F.; Beshay, S.; Bitar, M.; Kelly, J.; Syed, M.; Javitt, M. VIP in the Treatment of Critical COVID-19 Respiratory Failure in Patients with Severe Comorbidities. In Proceedings of the American Thoracic Society (ATS) 2021 International Conference, Virtual, 14–19 May 2021.
- Beshay, S.; Youssef, J.; Zahiruddin, F.; Al-Saadi, M.; Yau, S.; Goodarzi, A.; Huang, H.; Javitt, J. Rapid Clinical Recovery from Critical COVID-19 Pneumonia with Vasoactive Intestinal Peptide Treatment (Abstract 1268). J. Heart Lung Transpl. 2021, 40, S501.
- Youssef, J.G.; Javitt, J.; Lavin, P.; Al-Saadi, M.; Zahiruddin, F.; Beshay, S.; Bitar, M.; Kelley, J.; Sayed, M. VIP in the Treatment of Critical COVID-19 with Respiratory Failure in Patients with Severe Comorbidity: A Prospective Externally-Controlled Trial. Available online: https://ssrn.com/abstract=3665228 (accessed on 16 February 2022).