Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rabea Asleh | -- | 1929 | 2022-04-19 11:09:46 | | | |
| 2 | Catherine Yang | -1 word(s) | 1928 | 2022-04-19 11:21:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Asleh, R.; , .; Yarkoni, M. Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies. Encyclopedia. Available online: https://encyclopedia.pub/entry/21928 (accessed on 24 July 2026).
Asleh R, , Yarkoni M. Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies. Encyclopedia. Available at: https://encyclopedia.pub/entry/21928. Accessed July 24, 2026.
Asleh, Rabea, , Merav Yarkoni. "Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies" Encyclopedia, https://encyclopedia.pub/entry/21928 (accessed July 24, 2026).
Asleh, R., , ., & Yarkoni, M. (2022, April 19). Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies. In Encyclopedia. https://encyclopedia.pub/entry/21928
Asleh, Rabea, et al. "Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies." Encyclopedia. Web. 19 April, 2022.
Copy Citation
Cardiotoxicity ranges from arrhythmias to life-threatening myocarditis with very high mortality rates. To date, most treatments of Immune Checkpoint Inhibitor (ICI) cardiotoxicity include immune suppression, which is also not cardiac-specific and may result in hampering of tumor clearance.
immune checkpoint inhibitors
cardiotoxicity
lymphocyte
1. Cardiotoxicity of ICIs
The increased use of ICI therapy has revealed the extent and importance of ICI-induced cardiotoxicity (Figure 1). The severity of cardiac IRAEs is classified according to the American Society of Clinical Oncology [1] and ranges from benign ECG changes to life-threatening myocarditis. Recently, analysis of the Pharmacovigilance database and a meta-analysis of published papers and randomized controlled trials (RCTs) has identified cardiac events in 1.3–5.8% of patients receiving ICIs (single and dual therapy) [2]. Another large meta-analysis of RCTs showed increased odds ratios for developing myocarditis and pericarditis (4.4 and 2.2, respectively) in ICI-treated patients, with increased associated risks of heart failure and myocardial infarction (MI) [3]. Surprisingly, a meta-analysis of RCTs from phase II and III trials did not find any difference in CVAE between ICI-treated and control groups [4]. This may be explained by including highly selected patients in these trials (selection bias), reporting bias, and the type of analysis [3].
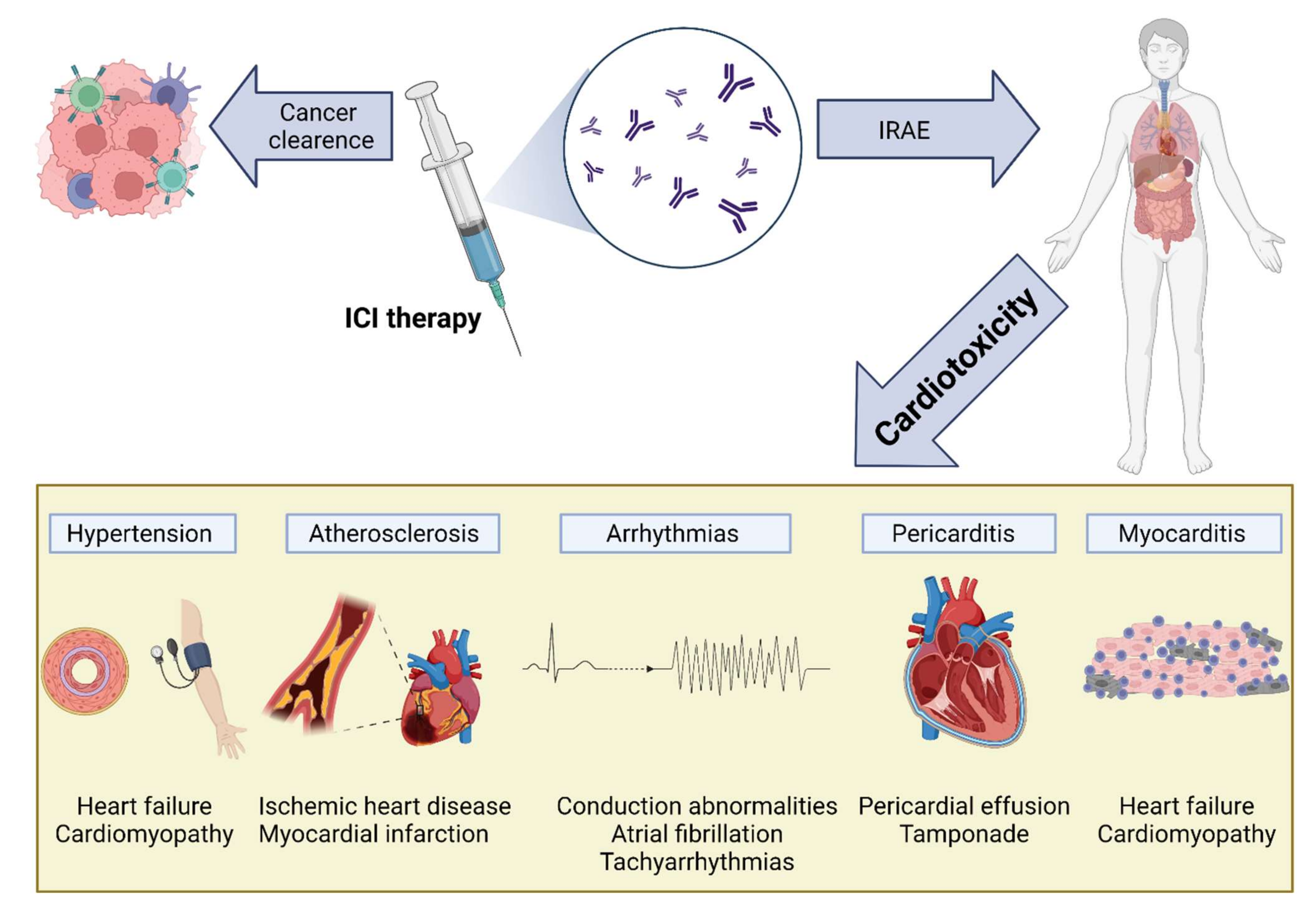
Figure 1. Cardiovascular toxicity induced by immune checkpoint inhibitor (ICI) therapy. The reactivation of T cells in response to ICIs is mediated by inflammation and fibrosis, leading to various cardiac manifestations, including myocarditis, pericarditis and pericardial effusion, arrhythmias, atherosclerosis, and hypertension: IRAEs, immune-related adverse events; ICI, immune checkpoint inhibitor.
The immune landscape of the resting heart is mostly dominated by macrophages [5][6], with low numbers of monocytes, dendritic cells, and mast cells present [7][8]. In addition, low numbers of T and B cells can be found [9][10]. Myocarditis, inflammation of the heart muscle, can be triggered by multiple etiologies and is characterized by lymphocyte-predominant infiltration (such as in viral myocarditis), eosinophilic infiltration (such as in hypereosinophilic syndrome), or the presence of giant cells (giant cell myocarditis). Like other IRAEs, ICI cardiotoxicity is also driven by lymphocyte, predominant immunity with CTLA4 and PD-L1 expression in the myocardium playing an important role in T-cell activity regulation [11][12][13][14].
2. Myocarditis
The most serious cardiac manifestation of ICI cardiotoxicity is the development of myocarditis. Although it may be silent with merely cardiac biomarker elevation, it may present as a fulminant disease, leading to cardiogenic shock and death [15]. A meta-analysis has reported that myocarditis may represent up to 50% of cardiac IRAEs (34 of 61 cardiac IRAEs among 4751 patients) [2], with mortality rates ranging from 20% to 50% [15][16][17][18][19]. Cardiac damage seems to start early after ICI treatment, with myocarditis reported to occur approximately 17 days (range: 13–64 days) following treatment initiation [20], but delayed appearance as far as 32 weeks after treatment initiation was reported [21]. PD-1 pathway inhibition seems to elicit more cardiotoxicity than CTLA4, and combinational therapy harbors the highest risk of myocarditis [18][20][22]. Interestingly, female patients are more likely to develop IRAEs and myocarditis [23][24]. This may be explained, at least partially, by PD-1 and PD-1L upregulation by estrogen [25][26], and is supported by an observation from CTLA4(+/−)/PD-1(−/−) deficient mice showing that female mice died at significantly higher rates than their male, age-matched counterparts [27].
Blockade of PD-1 and CTLA4 was shown to result in increased lymphocyte presence in the heart and other tissues [28]. This is supported by histological data from ICI-induced myocarditis patients showing increased CD4 and CD8 lymphocytes and, to a lesser degree, macrophages [20][29][30]. Interestingly, these T cells seem to have lower stimulatory thresholds for self-antigens [31]. Mouse models have shown that PD-1L is upregulated in endothelial and myocardial cells in response to inflammation or ischemia, presumably reducing the inflammatory response and tissue damage [20][32][33][34]. Several approaches have been perused to elucidate the mechanisms driving myocardial damage during ICI treatment. Initial mouse models showed varied effects, depending on the genetic background. PD-1 knockout in C57BL/6 mice displayed a normal phenotype [35], whereas deleting PD-1 in BALB/c background resulted in increased anti-troponin-I antibodies and dilated cardiomyopathy rather than myocarditis [11][35][36][37]. However, the transfer of CD8+ PD-1-deficient cell population to mice already experiencing myocarditis resulted in enhanced inflammation [38]. In MRL-lpr−/− mice (lacking FAS and predisposed to the development of systemic lupus erythematosus (SLE)-like phenotype), deletion of PD-1/PD-1L or the administration of neutralizing antibodies resulted in autoimmune myocarditis and CD4/8 infiltration; however, unlike in human ICI myocarditis, substantial levels of autoantibody formation against myosin were detected [27][39]. Interestingly, blocking PD-1 or CTLA4 in MRL mice results in increased lymphocyte infiltration without the development of overt myocarditis [27]. These differences between early mouse models and human ICI myocarditis may be explained by the fact that in mice most T cells are naïve, as opposed to most human T cells [27]. This notion is supported by studies examining myocarditis developing in melanoma mouse models treated with PD-1 blockade [14]. In contrast to solitary PD-1 deficiency, mouse models of CTLA-4 deficiency led to systemic inflammation involving T lymphocytes and resulted in early multiorgan failure and death [12][13]. This included myocarditis with CD8 cells infiltration [12][40]; however, the global activation of T lymphocytes also differs from ICI myocarditis. In light of these shortcomings of early mouse models, a primate model was developed showing CD4/CD8 T cell and low numbers of macrophage infiltration into cardiac tissues, with increased troponin-I and NT-proBNP levels [29]. Another approach to recapitulate ICI myocarditis in mice employed removing multiple participating genes. Deletion of both PD-1 and LAG-3 in BALB/c mice resulted in myocarditis with CD8 and CD4 cell infiltration, in addition to increased TNFα secretion [41][42]. More recently, a mouse model harboring a compound loss of CTLA4 and PD-1 was created. Ctla4+/− Pdcd1−/− mice showed increased troponin levels with cardiac infiltration of CD3, CD8, and CD4 cells, and reduced numbers of Treg cells, similar to that seen in human ICI myocarditis [27]. A more “physiologic” approach was employed by Lars et al., who injected immune-competent mice with melanoma cells, followed by anti-PD-1 antibodies treatment, resulting in functionally noticeable myocarditis with increased CD4 and CD8 cell infiltration [14]. Both Ctla4+/− Pdcd1−/− and melanoma models were used to assess treatment options (CTLA4-IgG1 and anti-TNFα, respectively) paving the way for a more comprehensive search for novel and more specific therapeutic options.
One reason for T-cell infiltration within the myocardium was elucidated by the recognition of similar clonal populations of T cells recognizing antigens expressed in both the TME and cardiac tissues [20]. Shared antigens between myeloma cells and cardiomyocytes have been later discovered [43]. This mechanism is further supported by the finding of similar TME and skin T cells in NSCLC presenting with skin IRAEs [44], suggesting a common underlying IRAE mechanism. In addition to antigen recognition, ICI treatment results in upregulation of CXCR3–CXCL9/CXCL10 and CCR5/CCL5 chemokines necessary for T-cell activation [29][45]. Following activation, T cells overexpress TNFα, INFγ, and granzyme B, thus promoting local cell damage and death [46][47][48]. This is also supported by in vitro co-culture experiments of cardiomyocytes exposed to lymphocytes and anti-PD-1/CTLA4 antibodies, demonstrating increased levels of leukotriene B4 [46]. The surprisingly high incidence of myasthenia gravis with ICI myocarditis suggests that, in addition to T cells, antibody-mediated damage might also contribute to ICI autoimmunity [49][50][51][52][53]. Antibodies against AChR and MuSK are commonly present (~66% (30 of 45 patients) and 5% (1 of 19 patients), respectively) in ICI-induced myasthenia gravis cases [54]. However, as mentioned, increased anti-troponin-I and anti-myosin antibodies seen in PD-1 mouse models result in dilated cardiomyopathy or myocarditis that differs from ICI myocarditis. Further investigation is needed to elucidate this aspect.
Taken together, myocarditis seems to develop by multiple steps including enhanced recruitment of T lymphocytes, reduced PD-1/PD-1L protection, reduced Treg activity, and T-cell recognition of shared epitopes. The development of novel mouse models will potentially allow better elucidation of other mechanisms driving ICI myocarditis and drive better-targeted treatment options.
3. Pericarditis
Pericardial effusion was reported following ICI treatment, with some developing overt pericarditis [55][56]. The incidence of pericardial effusion ranges from 0.3% (95/31321 cardiovascular IRAEs) but may be as high as 7% in NSCLC (4/60 patients receiving ICIs), and this has been associated with worse outcomes [17][57]. As pericardial effusion is present in multiple tumor types, it is important to differentiate the etiology driving pericardial effusion accumulation; however, the association of pericardial effusion with higher mortality in ICI-treated patients suggests a possible cardiotoxic effect. Indeed, pericardial samples from ICI-treated patients suffering from pericarditis showed significant lymphocytic infiltrates [58].
4. Arrhythmias
Atrial fibrillation (AF), conduction delays, and ventricular arrhythmias have all been observed following ICI treatment [15][20][31][59], and cardiac arrhythmias may represent a common cardiac IRAE [15][17][60][61] that may occur in up to 10% of patients receiving ICIs, according to one study [60]. This may represent isolated conduction toxicity or be part of a widespread manifestation of myocarditis [15]. Interestingly, CD4 PD-1 expression and PD-1L myeloid dendritic cells were found to be reduced in patients with AF [62]. Additionality, mouse models of myocarditis showed an increased incidence of arrhythmias [27]. The significantly worse outcome of patients presenting with conduction abnormalities (80% vs. 16% mortality) [15] following ICI treatment emphasizes its significance and the importance of ECG surveillance.
5. Atherosclerosis
Atherosclerosis has emerged as a new target of ICI-induced toxicity [63]. Indeed, acute coronary syndrome (ACS) incidence among ICI patients was reported to be around 0.5% (2/402 of ICI-treated patients) in a large metanalysis [64], but other reports of incidences as high as 2.4% (80/3326) to 3.6% (102/2842) of patients receiving ICI have emerged [19][65][66]. Although macrophages play major roles in the development of the atherosclerotic lesion, T lymphocytes were shown to occupy the atherosclerotic plaque [67] ], and their activation and secretion of INFγ lead to macrophage activation [68]. Interestingly, patients with coronary artery disease have reduced expression of PD-1 and PD-L1 on peripheral mononuclear cells [69]. The role of PD-1 in atherosclerosis is further supported by observations of larger atherosclerotic lesions in PD-1 knockout mice [70] with a more prominent T-cell response and larger necrotic cores [71]. As for CTLA-4, studies in mice have shown increased plaque size, following anti-CTLA-4 antibody treatment. Concurrently, treatment with abatacept (a CLTA-4 analog) or overexpression of CTLA-4 reduced plaque size [72][73][74]. The proatherosclerotic effect of PD-1 and CTLA4 inhibition seems to be, at least in part, mediated by the increase in INFγ and TNFα secretion, which are known mediators promoting coronary thrombosis [75]. As new immunomodulatory targets are discovered, it is interesting to see whether it can be possible to activate the immune response without increasing atherosclerotic risk [63].
Events of coronary spasm following ICI administration have been reported [76][77], but the full extent of this phenomenon warrants further investigation.
6. Emerging Treatment of Cardiac IRAEs
To date, the hallmark of IRAE management is based on steroid therapy and escalation to other immunosuppressive regiments in non-responders [1][78]. Immune suppression using the inosine-5′-monophosphate dehydrogenase (IMPDH) inhibitor, mycophenolate mofetil (MMF), results primarily in B- and T-cell inhibition [79][80]. Calcineurin inhibitors, such as tacrolimus, exert immunosuppressive properties through inhibiting T-cell activity [21][81][82][83]. Lymphopenia may be induced by antithymocyte globulin (ATG) [21][81][82], resulting in the cessation of autoimmunity. Other strategies include the use of intravenous immunoglobulin (IVIG) [49][50][51][84][85][86][87], and plasmapheresis [52][53][84][88][89][90] to incapacitate or remove ICIs (which have a long half-life of up to 27 days in some cases [59] can be employed.
Recent advances in the understanding of IRAE mechanisms have allowed the employment of targeted therapies. These include CTLA4-Ig (abatacept) to reimpose inhibition of activated T cells in myocarditis, as shown in mouse models [27] and case reports in humans [27][91]. Anti-CD52 (alemtuzumab), commonly used in CLL and multiple myeloma, has shown T-cell depletion capabilities and has been used in a case report with encouraging results [92]. Anti-TNFα (infliximab) has also shown promising results [1][14][84][93], although its utility in heart failure patients may be limited. It is worth noting that any treatment resulting in global quenching of the immune response also has the potential to allow tumor escape from immunosurveillance and disease progression. A better understanding of both the stimulatory and inhibitory molecules implicated in intracellular signaling pathways may allow better control of adverse events while allowing the tenacious antitumoral activity.
References
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714–1768.
- Rubio-Infante, N.; Ramírez-Flores, Y.A.; Castillo, E.C.; Lozano, O.; García-Rivas, G.; Torre-Amione, G. Cardiotoxicity associated with immune checkpoint inhibitor therapy: A meta-analysis. Eur. J. Heart Fail. 2021, 23, 1739–1747.
- Dolladille, C.; Akroun, J.; Morice, P.-M.; Dompmartin, A.; Ezine, E.; Sassier, M.; Da-Silva, A.; Plane, A.-F.; Legallois, D.; L’Orphelin, J.-M.; et al. Cardiovascular immunotoxicities associated with immune checkpoint inhibitors: A safety meta-analysis. Eur. Heart J. 2021, 42, 4964–4977.
- Agostinetto, E.; Eiger, D.; Lambertini, M.; Ceppi, M.; Bruzzone, M.; Pondé, N.; Plummer, C.; Awada, A.H.; Santoro, A.; Piccart-Gebhart, M.; et al. Cardiotoxicity of immune checkpoint inhibitors: A systematic review and meta-analysis of randomised clinical trials. Eur. J. Cancer 2021, 148, 76–91.
- Pinto, A.R.; Paolicelli, R.; Salimova, E.; Gospocic, J.; Slonimsky, E.; Bilbao-Cortes, D.; Godwin, J.W.; Rosenthal, N.A. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS ONE 2012, 7, e36814.
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047.
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710.
- Choi, J.-H.; Do, Y.; Cheong, C.; Koh, H.; Boscardin, S.B.; Oh, Y.-S.; Bozzacco, L.; Trumpfheller, C.; Park, C.G.; Steinman, R.M. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J. Exp. Med. 2009, 206, 497–505.
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280.
- Saxena, A.; Dobaczewski, M.; Rai, V.; Haque, Z.; Chen, W.; Li, N.; Frangogiannis, N.G. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1233-42.
- Tarrio, M.L.; Grabie, N.; Bu, D.; Sharpe, A.H.; Lichtman, A.H. PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J. Immunol. 2012, 188, 4876–4884.
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547.
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988.
- Michel, L.; Helfrich, I.; Hendgen-Cotta, U.B.; Mincu, R.-I.; Korste, S.; Mrotzek, S.M.; Spomer, A.; Odersky, A.; Rischpler, C.; Herrmann, K.; et al. Targeting early stages of cardiotoxicity from anti-PD1 immune checkpoint inhibitor therapy. Eur. Heart J. 2021, 14, 316–329.
- Escudier, M.; Cautela, J.; Malissen, N.; Ancedy, Y.; Orabona, M.; Pinto, J.; Monestier, S.; Grob, J.-J.; Scemama, U.; Jacquier, A.; et al. Clinical Features, Management, and Outcomes of Immune Checkpoint Inhibitor-Related Cardiotoxicity. Circulation 2017, 136, 2085–2087.
- Moslehi, J.J.; Salem, J.-E.; Sosman, J.A.; Lebrun-Vignes, B.; Johnson, D.B. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet 2018, 391, 933.
- Salem, J.-E.; Manouchehri, A.; Moey, M.; Lebrun-Vignes, B.; Bastarache, L.; Pariente, A.; Gobert, A.; Spano, J.-P.; Balko, J.M.; Bonaca, M.P.; et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: An observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018, 19, 1579–1589.
- Mahmood, S.S.; Fradley, M.G.; Cohen, J.V.; Nohria, A.; Reynolds, K.L.; Heinzerling, L.M.; Sullivan, R.J.; Damrongwatanasuk, R.; Chen, C.L.; Gupta, D.; et al. Myocarditis in Patients Treated with Immune Checkpoint Inhibitors. J. Am. Coll. Cardiol. 2018, 71, 1755–1764.
- Oren, O.; Yang, E.H.; Molina, J.R.; Bailey, K.R.; Blumenthal, R.S.; Kopecky, S.L. Cardiovascular Health and Outcomes in Cancer Patients Receiving Immune Checkpoint Inhibitors. Am. J. Cardiol. 2020, 125, 1920–1926.
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N. Engl. J. Med. 2016, 375, 1749–1755.
- Jain, V.; Bahia, J.; Mohebtash, M.; Barac, A. Cardiovascular Complications Associated with Novel Cancer Immunotherapies. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 36.
- Zimmer, L.; Goldinger, S.M.; Hofmann, L.; Loquai, C.; Ugurel, S.; Thomas, I.; Schmidgen, M.I.; Gutzmer, R.; Utikal, J.S.; Göppner, D.; et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur. J. Cancer 2016, 60, 210–225.
- Zamami, Y.; Niimura, T.; Okada, N.; Koyama, T.; Fukushima, K.; Izawa-Ishizawa, Y.; Ishizawa, K. Factors Associated With Immune Checkpoint Inhibitor-Related Myocarditis. JAMA Oncol. 2019, 5, 1635–1637.
- Valpione, S.; Pasquali, S.; Campana, L.G.; Piccin, L.; Mocellin, S.; Pigozzo, J.; Chiarion-Sileni, V. Sex and interleukin-6 are prognostic factors for autoimmune toxicity following treatment with anti-CTLA4 blockade. J. Transl. Med. 2018, 16, 94.
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J. Neurosci. Res. 2006, 84, 370–378.
- Wang, C.; Dehghani, B.; Li, Y.; Kaler, L.J.; Vandenbark, A.A.; Offner, H. Oestrogen modulates experimental autoimmune encephalomyelitis and interleukin-17 production via programmed death 1. Immunology 2009, 126, 329–335.
- Wei, S.C.; Meijers, W.C.; Axelrod, M.L.; Anang, N.-A.A.S.; Screever, E.M.; Wescott, E.C.; Johnson, D.B.; Whitley, E.; Lehmann, L.; Courand, P.Y.; et al. A Genetic Mouse Model Recapitulates Immune Checkpoint Inhibitor-Associated Myocarditis and Supports a Mechanism-Based Therapeutic Intervention. Cancer Discov. 2021, 11, 614–625.
- Weinmann, S.C.; Pisetsky, D.S. Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatology 2019, 58 (Suppl. S7), vii59–vii67.
- Ji, C.; Roy, M.D.; Golas, J.; Vitsky, A.; Ram, S.; Kumpf, S.W.; Martin, M.; Barletta, F.; Meier, W.A.; Hooper, A.T.; et al. Myocarditis in Cynomolgus Monkeys Following Treatment with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2019, 25, 4735–4748.
- Ganatra, S.; Neilan, T.G. Immune Checkpoint Inhibitor-Associated Myocarditis. Oncologist 2018, 23, 879–886.
- Heinzerling, L.; Ott, P.A.; Hodi, F.S.; Husain, A.N.; Tajmir-Riahi, A.; Tawbi, H.; Pauschinger, M.; Gajewski, T.F.; Lipson, E.J.; Luke, J.J. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J. Immunother. Cancer 2016, 4, 50.
- Grabie, N.; Gotsman, I.; DaCosta, R.; Pang, H.; Stavrakis, G.; Butte, M.J.; Keir, M.E.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation 2007, 116, 2062–2071.
- Proietti, I.; Skroza, N.; Michelini, S.; Mambrin, A.; Balduzzi, V.; Bernardini, N.; Martin, M.; Barletta, F.; Meier, W.A.; Hooper, A.T.; et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers 2020, 12, 1823.
- Keir, M.E.; Freeman, G.J.; Sharpe, A.H. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J. Immunol. 2007, 179, 5064–5070.
- Okazaki, T.; Tanaka, Y.; Nishio, R.; Mitsuiye, T.; Mizoguchi, A.; Wang, J.; Ishida, M.; Hiai, H.; Matsumori, A.; Minato, N.; et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 2003, 9, 1477–1483.
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, A.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322.
- Parmacek, M.S.; Solaro, R.J. Biology of the troponin complex in cardiac myocytes. Prog. Cardiovasc. Dis. 2004, 47, 159–176.
- Lucas, J.A.; Menke, J.; Rabacal, W.A.; Schoen, F.J.; Sharpe, A.H.; Kelley, V.R. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J. Immunol. 2008, 181, 2513–2521.
- Wang, J.; Okazaki, I.-M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 2010, 22, 443–452.
- Love, V.A.; Grabie, N.; Duramad, P.; Stavrakis, G.; Sharpe, A.; Lichtman, A. CTLA-4 ablation and interleukin-12 driven differentiation synergistically augment cardiac pathogenicity of cytotoxic T lymphocytes. Circ. Res. 2007, 101, 248–257.
- Okazaki, T.; Okazaki, I.; Wang, J.; Sugiura, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T.; et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011, 208, 395–407.
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927.
- Martinez-Calle, N.; Rodriguez-Otero, P.; Villar, S.; Mejías, L.; Melero, I.; Prosper, F.; Marinello, P.; Paiva, B.; Idoate, M.; San-Miguel, J. Anti-PD1 associated fulminant myocarditis after a single pembrolizumab dose: The role of occult pre-existing autoimmunity. Haematologica 2018, 103, e318–e321.
- Berner, F.; Bomze, D.; Diem, S.; Ali, O.H.; Fässler, M.; Ring, S.; Niederer, R.; Ackermann, C.J.; Baumgaertner, P.; Pikor, N.; et al. Association of Checkpoint Inhibitor-Induced Toxic Effects With Shared Cancer and Tissue Antigens in Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 1043–1047.
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; Baba, M.H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47.
- Quagliariello, V.; Passariello, M.; Rea, D.; Barbieri, A.; Iovine, M.; Bonelli, A.; Caronna, A.; Botti, G.; Lorenzo, C.D.; Maurea, N. Evidences of CTLA-4 and PD-1 Blocking Agents-Induced Cardiotoxicity in Cellular and Preclinical Models. J. Pers. Med. 2020, 10, 179.
- Tocchetti, C.G.; Galdiero, M.R.; Varricchi, G. Cardiac Toxicity in Patients Treated with Immune Checkpoint Inhibitors: It Is Now Time for Cardio-Immuno-Oncology. J. Am. Coll. Cardiol. 2018, 71, 1765–1767.
- Varricchi, G.; Galdiero, M.R.; Marone, G.; Criscuolo, G.; Triassi, M.; Bonaduce, D.; Marone, G.; Tocchetti, C.G. Cardiotoxicity of immune checkpoint inhibitors. ESMO Open 2017, 2, e000247.
- Xing, Q.; Zhang, Z.-W.; Lin, Q.-H.; Shen, L.-H.; Wang, P.-M.; Zhang, S.; Fan, M.; Zhu, B. Myositis-myasthenia gravis overlap syndrome complicated with myasthenia crisis and myocarditis associated with anti-programmed cell death-1 (sintilimab) therapy for lung adenocarcinoma. Ann. Transl. Med. 2020, 8, 250.
- Yanase, T.; Moritoki, Y.; Kondo, H.; Ueyama, D.; Akita, H.; Yasui, T. Myocarditis and myasthenia gravis by combined nivolumab and ipilimumab immunotherapy for renal cell carcinoma: A case report of successful management. Urol. Case Rep. 2021, 34, 101508.
- Fazel, M.; Jedlowski, P.M. Severe Myositis, Myocarditis, and Myasthenia Gravis with Elevated Anti-Striated Muscle Antibody following Single Dose of Ipilimumab-Nivolumab Therapy in a Patient with Metastatic Melanoma. Case Rep. Immunol. 2019, 2019, 2539493.
- Kimura, T.; Fukushima, S.; Miyashita, A.; Aoi, J.; Jinnin, M.; Kosaka, T.; Ando, Y.; Matsukawa, M.; Inoue, H.; Kiyotani, K.; et al. Myasthenic crisis and polymyositis induced by one dose of nivolumab. Cancer Sci. 2016, 107, 1055–1058.
- Rota, E.; Varese, P.; Agosti, S.; Celli, L.; Ghiglione, E.; Pappalardo, I.; Zaccone, G.; Paglia, A.; Morelli, N. Concomitant myasthenia gravis, myositis, myocarditis and polyneuropathy, induced by immune-checkpoint inhibitors: A life-threatening continuum of neuromuscular and cardiac toxicity. eNeurologicalSci 2019, 14, 4–5.
- Huang, Y.-T.; Chen, Y.-P.; Lin, W.-C.; Su, W.-C.; Sun, Y.-T. Immune Checkpoint Inhibitor-Induced Myasthenia Gravis. Front. Neurol. 2020, 11, 634.
- Palaskas, N.; Morgan, J.; Daigle, T.; Banchs, J.; Durand, J.-B.; Hong, D.; Naing, A.; Le, H.; Hassan, S.A.; Karimzad, K.; et al. Targeted Cancer Therapies With Pericardial Effusions Requiring Pericardiocentesis Focusing on Immune Checkpoint Inhibitors. Am. J. Cardiol. 2019, 123, 1351–1357.
- Kolla, B.C.; Patel, M.R. Recurrent pleural effusions and cardiac tamponade as possible manifestations of pseudoprogression associated with nivolumab therapy—A report of two cases. J. Immunother. Cancer 2016, 4, 80.
- Canale, M.L.; Camerini, A.; Casolo, G.; Lilli, A.; Bisceglia, I.; Parrini, I.; Lestuzzi, C.; Meglio, J.D.; Puccetti, C.; Camerini, L.; et al. Incidence of Pericardial Effusion in Patients with Advanced Non-Small Cell Lung Cancer Receiving Immunotherapy. Adv. Ther. 2020, 37, 3178–3184.
- Altan, M.; Toki, M.I.; Gettinger, S.N.; Carvajal-Hausdorf, D.E.; Zugazagoitia, J.; Sinard, J.H.; Herbst, R.S.; Rimm, D.L. Immune Checkpoint Inhibitor-Associated Pericarditis. J. Thorac. Oncol. 2019, 14, 1102–1108.
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502.
- Mir, H.; Alhussein, M.; Alrashidi, S.; Alzayer, H.; Alshatti, A.; Valettas, N.; Mukherjee, S.D.; Nair, V.; Leong, D.P. Cardiac Complications Associated With Checkpoint Inhibition: A Systematic Review of the Literature in an Important Emerging Area. Can. J. Cardiol. 2018, 34, 1059–1068.
- D’Souza, M.; Nielsen, D.; Svane, I.M.; Iversen, K.; Rasmussen, P.V.; Madelaire, C.; Fosbøl, E.; Køber, L.; Gustafsson, F.; Andersson, C.; et al. The risk of cardiac events in patients receiving immune checkpoint inhibitors: A nationwide Danish study. Eur. Heart J. 2021, 42, 1621–1631.
- Liu, L.; Zheng, Q.; Lee, J.; Ma, Z.; Zhu, Q.; Wang, Z. PD-1/PD-L1 expression on CD(4+) T cells and myeloid DCs correlates with the immune pathogenesis of atrial fibrillation. J. Cell. Mol. Med. 2015, 19, 1223–1233.
- Vuong, J.T.; Stein-Merlob, A.F.; Nayeri, A.; Sallam, T.; Neilan, T.G.; Yang, E.H. Immune Checkpoint Therapies and Atherosclerosis: Mechanisms and Clinical Implications: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 577–593.
- Hu, Y.-B.; Zhang, Q.; Li, H.-J.; Michot, J.M.; Liu, H.-B.; Zhan, P.; Lv, T.-F.; Song, Y. Evaluation of rare but severe immune related adverse effects in PD-1 and PD-L1 inhibitors in non-small cell lung cancer: A meta-analysis. Transl. Lung Cancer Res. 2017, 6 (Suppl. S1), S8–S20.
- Cautela, J.; Rouby, F.; Salem, J.-E.; Alexandre, J.; Scemama, U.; Dolladille, C.; Cohen, A.; Paganelli, F.; Ederhy, S.; Thuny, F. Acute Coronary Syndrome With Immune Checkpoint Inhibitors: A Proof-of-Concept Case and Pharmacovigilance Analysis of a Life-Threatening Adverse Event. Can. J. Cardiol. 2020, 36, 476–481.
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association Between Immune Checkpoint Inhibitors With Cardiovascular Events and Atherosclerotic Plaque. Circulation 2020, 142, 2299–2311.
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401.
- Stemme, S.; Faber, B.; Holm, J.; Wiklund, O.; Witztum, J.L.; Hansson, G.K. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1995, 92, 3893–3897.
- Lee, J.; Zhuang, Y.; Wei, X.; Shang, F.; Wang, J.; Zhang, Y.; Liu, X.; Yang, Y.; Liu, L.; Zheng, Q. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J. Mol. Cell. Cardiol. 2009, 46, 169–176.
- Bu, D.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1100–1107.
- Poels, K.; van Leent, M.M.T.; Boutros, C.; Tissot, H.; Roy, S.; Meerwaldt, A.E.; Toner, Y.C.A.; Reiche, M.E.; Kusters, P.J.H.; Malinova, T.; et al. Immune Checkpoint Inhibitor Therapy Aggravates T Cell-Driven Plaque Inflammation in Atherosclerosis. JACC CardioOncol. 2020, 2, 599–610.
- Ewing, M.M.; Karper, J.C.; Abdul, S.; de Jong, R.C.M.; Peters, H.A.B.; de Vries, M.R.; Redeker, A.; Kuiper, J.; Toes, R.E.M.; Arens, R.; et al. T-cell co-stimulation by CD28-CD80/86 and its negative regulator CTLA-4 strongly influence accelerated atherosclerosis development. Int. J. Cardiol. 2013, 168, 1965–1974.
- Poels, K.; van Leent, M.M.T.; Reiche, M.E.; Kusters, P.J.H.; Huveneers, S.; de Winther, M.P.J.; Mulder, W.J.M.; Lutgens, E.; Seijkens, T.T.P. Antibody-Mediated Inhibition of CTLA4 Aggravates Atherosclerotic Plaque Inflammation and Progression in Hyperlipidemic Mice. Cells 2020, 9, 1987.
- Matsumoto, T.; Sasaki, N.; Yamashita, T.; Emoto, T.; Kasahara, K.; Mizoguchi, T.; Hayashi, T.; Yodoi, K.; Kitano, N.; Saito, T.; et al. Overexpression of Cytotoxic T-Lymphocyte-Associated Antigen-4 Prevents Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1141–1151.
- Foks, A.C.; Kuiper, J. Immune checkpoint proteins: Exploring their therapeutic potential to regulate atherosclerosis. Br. J. Pharmacol. 2017, 174, 3940–3955.
- Nykl, R.; Fischer, O.; Vykoupil, K.; Taborsky, M. A unique reason for coronary spasm causing temporary ST elevation myocardial infarction (inferior STEMI)—systemic inflammatory response syndrome after use of pembrolizumab. Arch. Med. Sci. Atheroscler. Dis. 2017, 2, e100–e102.
- Otsu, K.; Tajiri, K.; Sakai, S.; Ieda, M. Vasospastic angina following immune checkpoint blockade. Eur. Heart J. 2020, 41, 1702.
- Schneider, B.J.; Naidoo, J.; Santomasso, B.D.; Lacchetti, C.; Adkins, S.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 4073–4126.
- Reddy, N.; Moudgil, R.; Lopez-Mattei, J.C.; Karimzad, K.; Mouhayar, E.N.; Somaiah, N.; Conley, A.P.; Patel, S.; Giza, D.E.; Iliescu, C. Progressive and Reversible Conduction Disease With Checkpoint Inhibitors. Can. J. Cardiol. 2017, 33, 1335.e13–1335.e15.
- Hu, J.-R.; Florido, R.; Lipson, E.J.; Naidoo, J.; Ardehali, R.; Tocchetti, C.G.; Lyon, A.R.; Padera, R.F.; Johnson, D.B.; Moslehi, J. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc. Res. 2019, 115, 854–868.
- Tay, R.Y.; Blackley, E.; McLean, C.; Moore, M.; Bergin, P.; Gill, S.; Haydon, A. Successful use of equine anti-thymocyte globulin (ATGAM) for fulminant myocarditis secondary to nivolumab therapy. Br. J. Cancer 2017, 117, 921–924.
- Jain, V.; Mohebtash, M.; Rodrigo, M.E.; Ruiz, G.; Atkins, M.B.; Barac, A. Autoimmune Myocarditis Caused by Immune Checkpoint Inhibitors Treated with Antithymocyte Globulin. J. Immunother. 2018, 41, 332–335.
- Wang, D.Y.; Okoye, G.D.; Neilan, T.G.; Johnson, D.B.; Moslehi, J.J. Cardiovascular Toxicities Associated with Cancer Immunotherapies. Curr. Cardiol. Rep. 2017, 19, 21.
- Frigeri, M.; Meyer, P.; Banfi, C.; Giraud, R.; Hachulla, A.-L.; Spoerl, D.; Friedlaender, A.; Pugliesi-Rinaldi, A.; Dietrich, P.-Y. Immune Checkpoint Inhibitor-Associated Myocarditis: A New Challenge for Cardiologists. Can. J. Cardiol. 2018, 34, 92.e1–92.e3.
- Caforio, A.L.P.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648, 2648a–2648d.
- Balanescu, D.V.; Donisan, T.; Palaskas, N.; Lopez-Mattei, J.; Kim, P.Y.; Buja, L.M.; McNamara, D.M.; Kobashigawa, J.A.; Durand, J.-B.; Iliescu, C.A. Immunomodulatory treatment of immune checkpoint inhibitor-induced myocarditis: Pathway toward precision-based therapy. Cardiovasc. Pathol. 2020, 47, 107211.
- Yamaguchi, S.; Morimoto, R.; Okumura, T.; Yamashita, Y.; Haga, T.; Kuwayama, T.; Yokoi, T.; Hiraiwa, H.; Kondo, T.; Sugiura, Y.; et al. Late-Onset Fulminant Myocarditis With Immune Checkpoint Inhibitor Nivolumab. Can. J. Cardiol. 2018, 34, e1–e812.
- Compton, F.; He, L.; Sarode, R.; Wodajo, A.; Usmani, A.; Burner, J.; Berlacher, M.; Simone, N.D. Immune checkpoint inhibitor toxicity: A new indication for therapeutic plasma exchange? J. Clin. Apher. 2021, 36, 645–648.
- Schiopu, S.R.I.; Käsmann, L.; Schönermarck, U.; Fischereder, M.; Grabmaier, U.; Manapov, F.; Rauch, J.; Orban, M. Pembrolizumab-induced myocarditis in a patient with malignant mesothelioma: Plasma exchange as a successful emerging therapy-case report. Transl. Lung Cancer Res. 2021, 10, 1039–1046.
- Yogasundaram, H.; Alhumaid, W.; Chen, J.W.; Church, M.; Alhulaimi, N.; Kimber, S.; Paterson, D.I.; Senaratne, J.M. Plasma Exchange for Immune Checkpoint Inhibitor-Induced Myocarditis. CJC Open 2021, 3, 379–382.
- Salem, J.-E.; Allenbach, Y.; Vozy, A.; Brechot, N.; Johnson, D.B.; Moslehi, J.J.; Kerneis, M. Abatacept for Severe Immune Checkpoint Inhibitor-Associated Myocarditis. N. Engl. J. Med. 2019, 380, 2377–2379.
- Esfahani, K.; Buhlaiga, N.; Thébault, P.; Lapointe, R.; Johnson, N.A.; Miller, W.H. Alemtuzumab for Immune-Related Myocarditis Due to PD-1 Therapy. N. Engl. J. Med. 2019, 380, 2375–2376.
- Lyon, A.R.; Yousaf, N.; Battisti, N.M.L.; Moslehi, J.; Larkin, J. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. 2018, 19, E447–E458.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
960
Revisions:
2 times
(View History)
Update Date:
19 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No