Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
Cardiotoxicity ranges from arrhythmias to life-threatening myocarditis with very high mortality rates. To date, most treatments of Immune Checkpoint Inhibitor (ICI) cardiotoxicity include immune suppression, which is also not cardiac-specific and may result in hampering of tumor clearance.
- immune checkpoint inhibitors
- cardiotoxicity
- lymphocyte
1. Cardiotoxicity of ICIs
The increased use of ICI therapy has revealed the extent and importance of ICI-induced cardiotoxicity (Figure 4). The severity of cardiac IRAEs is classified according to the American Society of Clinical Oncology [153] and ranges from benign ECG changes to life-threatening myocarditis. Recently, analysis of the Pharmacovigilance database and a meta-analysis of published papers and randomized controlled trials (RCTs) has identified cardiac events in 1.3–5.8% of patients receiving ICIs (single and dual therapy) [154]. Another large meta-analysis of RCTs showed increased odds ratios for developing myocarditis and pericarditis (4.4 and 2.2, respectively) in ICI-treated patients, with increased associated risks of heart failure and myocardial infarction (MI) [155]. Surprisingly, a meta-analysis of RCTs from phase II and III trials did not find any difference in CVAE between ICI-treated and control groups [156]. This may be explained by including highly selected patients in these trials (selection bias), reporting bias, and the type of analysis [155].
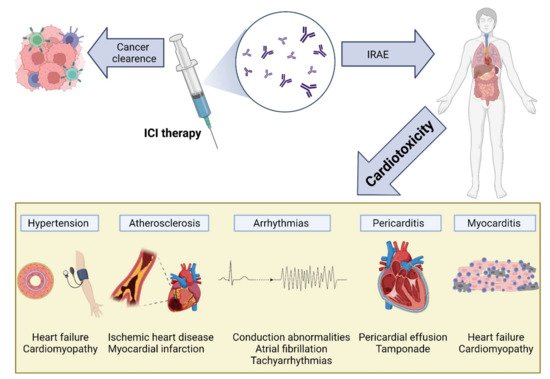
Figure 4. Cardiovascular toxicity induced by immune checkpoint inhibitor (ICI) therapy. The reactivation of T cells in response to ICIs is mediated by inflammation and fibrosis, leading to various cardiac manifestations, including myocarditis, pericarditis and pericardial effusion, arrhythmias, atherosclerosis, and hypertension: IRAEs, immune-related adverse events; ICI, immune checkpoint inhibitor.
The immune landscape of the resting heart is mostly dominated by macrophages [157,158], with low numbers of monocytes, dendritic cells, and mast cells present [159,160]. In addition, low numbers of T and B cells can be found [161,162]. Myocarditis, inflammation of the heart muscle, can be triggered by multiple etiologies and is characterized by lymphocyte-predominant infiltration (such as in viral myocarditis), eosinophilic infiltration (such as in hypereosinophilic syndrome), or the presence of giant cells (giant cell myocarditis). Like other IRAEs, ICI cardiotoxicity is also driven by lymphocyte, predominant immunity with CTLA4 and PD-L1 expression in the myocardium playing an important role in T-cell activity regulation [163,164,165,166].
2. Myocarditis
The most serious cardiac manifestation of ICI cardiotoxicity is the development of myocarditis. Although it may be silent with merely cardiac biomarker elevation, it may present as a fulminant disease, leading to cardiogenic shock and death [11]. A meta-analysis has reported that myocarditis may represent up to 50% of cardiac IRAEs (34 of 61 cardiac IRAEs among 4751 patients) [154], with mortality rates ranging from 20% to 50% [5,6,10,11,14]. Cardiac damage seems to start early after ICI treatment, with myocarditis reported to occur approximately 17 days (range: 13–64 days) following treatment initiation [4], but delayed appearance as far as 32 weeks after treatment initiation was reported [167]. PD-1 pathway inhibition seems to elicit more cardiotoxicity than CTLA4, and combinational therapy harbors the highest risk of myocarditis [4,10,168]. Interestingly, female patients are more likely to develop IRAEs and myocarditis [169,170]. This may be explained, at least partially, by PD-1 and PD-1L upregulation by estrogen [171,172], and is supported by an observation from CTLA4(+/−)/PD-1(−/−) deficient mice showing that female mice died at significantly higher rates than their male, age-matched counterparts [173].
Blockade of PD-1 and CTLA4 was shown to result in increased lymphocyte presence in the heart and other tissues [174]. This is supported by histological data from ICI-induced myocarditis patients showing increased CD4 and CD8 lymphocytes and, to a lesser degree, macrophages [4,175,176]. Interestingly, these T cells seem to have lower stimulatory thresholds for self-antigens [139]. Mouse models have shown that PD-1L is upregulated in endothelial and myocardial cells in response to inflammation or ischemia, presumably reducing the inflammatory response and tissue damage [4,177,178,179]. Several approaches have been perused to elucidate the mechanisms driving myocardial damage during ICI treatment. Initial mouse models showed varied effects, depending on the genetic background. PD-1 knockout in C57BL/6 mice displayed a normal phenotype [180], whereas deleting PD-1 in BALB/c background resulted in increased anti-troponin-I antibodies and dilated cardiomyopathy rather than myocarditis [143,163,180,181]. However, the transfer of CD8+ PD-1-deficient cell population to mice already experiencing myocarditis resulted in enhanced inflammation [182]. In MRL-lpr−/− mice (lacking FAS and predisposed to the development of systemic lupus erythematosus (SLE)-like phenotype), deletion of PD-1/PD-1L or the administration of neutralizing antibodies resulted in autoimmune myocarditis and CD4/8 infiltration; however, unlike in human ICI myocarditis, substantial levels of autoantibody formation against myosin were detected [173,183]. Interestingly, blocking PD-1 or CTLA4 in MRL mice results in increased lymphocyte infiltration without the development of overt myocarditis [173]. These differences between early mouse models and human ICI myocarditis may be explained by the fact that in mice most T cells are naïve, as opposed to most human T cells [173]. This notion is supported by studies examining myocarditis developing in melanoma mouse models treated with PD-1 blockade [166]. In contrast to solitary PD-1 deficiency, mouse models of CTLA-4 deficiency led to systemic inflammation involving T lymphocytes and resulted in early multiorgan failure and death [164,165]. This included myocarditis with CD8 cells infiltration [164,184]; however, the global activation of T lymphocytes also differs from ICI myocarditis. In light of these shortcomings of early mouse models, a primate model was developed showing CD4/CD8 T cell and low numbers of macrophage infiltration into cardiac tissues, with increased troponin-I and NT-proBNP levels [175]. Another approach to recapitulate ICI myocarditis in mice employed removing multiple participating genes. Deletion of both PD-1 and LAG-3 in BALB/c mice resulted in myocarditis with CD8 and CD4 cell infiltration, in addition to increased TNFα secretion [185,186]. More recently, a mouse model harboring a compound loss of CTLA4 and PD-1 was created. Ctla4+/− Pdcd1−/− mice showed increased troponin levels with cardiac infiltration of CD3, CD8, and CD4 cells, and reduced numbers of Treg cells, similar to that seen in human ICI myocarditis [173]. A more “physiologic” approach was employed by Lars et al., who injected immune-competent mice with melanoma cells, followed by anti-PD-1 antibodies treatment, resulting in functionally noticeable myocarditis with increased CD4 and CD8 cell infiltration [166]. Both Ctla4+/− Pdcd1−/− and melanoma models were used to assess treatment options (CTLA4-IgG1 and anti-TNFα, respectively) paving the way for a more comprehensive search for novel and more specific therapeutic options.
One reason for T-cell infiltration within the myocardium was elucidated by the recognition of similar clonal populations of T cells recognizing antigens expressed in both the TME and cardiac tissues [4]. Shared antigens between myeloma cells and cardiomyocytes have been later discovered [144]. This mechanism is further supported by the finding of similar TME and skin T cells in NSCLC presenting with skin IRAEs [142], suggesting a common underlying IRAE mechanism. In addition to antigen recognition, ICI treatment results in upregulation of CXCR3–CXCL9/CXCL10 and CCR5/CCL5 chemokines necessary for T-cell activation [175,187]. Following activation, T cells overexpress TNFα, INFγ, and granzyme B, thus promoting local cell damage and death [188,189,190]. This is also supported by in vitro co-culture experiments of cardiomyocytes exposed to lymphocytes and anti-PD-1/CTLA4 antibodies, demonstrating increased levels of leukotriene B4 [188]. The surprisingly high incidence of myasthenia gravis with ICI myocarditis suggests that, in addition to T cells, antibody-mediated damage might also contribute to ICI autoimmunity [191,192,193,194,195]. Antibodies against AChR and MuSK are commonly present (~66% (30 of 45 patients) and 5% (1 of 19 patients), respectively) in ICI-induced myasthenia gravis cases [196]. However, as mentioned, increased anti-troponin-I and anti-myosin antibodies seen in PD-1 mouse models result in dilated cardiomyopathy or myocarditis that differs from ICI myocarditis. Further investigation is needed to elucidate this aspect.
Taken together, myocarditis seems to develop by multiple steps including enhanced recruitment of T lymphocytes, reduced PD-1/PD-1L protection, reduced Treg activity, and T-cell recognition of shared epitopes. The development of novel mouse models will potentially allow better elucidation of other mechanisms driving ICI myocarditis and drive better-targeted treatment options (Figure 3).
3. Pericarditis
Pericardial effusion was reported following ICI treatment, with some developing overt pericarditis [197,198]. The incidence of pericardial effusion ranges from 0.3% (95/31321 cardiovascular IRAEs) but may be as high as 7% in NSCLC (4/60 patients receiving ICIs), and this has been associated with worse outcomes [6,199]. As pericardial effusion is present in multiple tumor types, it is important to differentiate the etiology driving pericardial effusion accumulation; however, the association of pericardial effusion with higher mortality in ICI-treated patients suggests a possible cardiotoxic effect. Indeed, pericardial samples from ICI-treated patients suffering from pericarditis showed significant lymphocytic infiltrates [200].
4. Arrhythmias
Atrial fibrillation (AF), conduction delays, and ventricular arrhythmias have all been observed following ICI treatment [4,11,139,201], and cardiac arrhythmias may represent a common cardiac IRAE [6,11,202,203] that may occur in up to 10% of patients receiving ICIs, according to one study [202]. This may represent isolated conduction toxicity or be part of a widespread manifestation of myocarditis [11]. Interestingly, CD4 PD-1 expression and PD-1L myeloid dendritic cells were found to be reduced in patients with AF [204]. Additionality, mouse models of myocarditis showed an increased incidence of arrhythmias [173]. The significantly worse outcome of patients presenting with conduction abnormalities (80% vs. 16% mortality) [11] following ICI treatment emphasizes its significance and the importance of ECG surveillance.
5. Atherosclerosis
Atherosclerosis has emerged as a new target of ICI-induced toxicity [205]. Indeed, acute coronary syndrome (ACS) incidence among ICI patients was reported to be around 0.5% (2/402 of ICI-treated patients) in a large metanalysis [206], but other reports of incidences as high as 2.4% (80/3326) to 3.6% (102/2842) of patients receiving ICI have emerged [14,207,208]. Although macrophages play major roles in the development of the atherosclerotic lesion, T lymphocytes were shown to occupy the atherosclerotic plaque [209] ], and their activation and secretion of INFγ lead to macrophage activation [210]. Interestingly, patients with coronary artery disease have reduced expression of PD-1 and PD-L1 on peripheral mononuclear cells [211]. The role of PD-1 in atherosclerosis is further supported by observations of larger atherosclerotic lesions in PD-1 knockout mice [212] with a more prominent T-cell response and larger necrotic cores [213]. As for CTLA-4, studies in mice have shown increased plaque size, following anti-CTLA-4 antibody treatment. Concurrently, treatment with abatacept (a CLTA-4 analog) or overexpression of CTLA-4 reduced plaque size [214,215,216]. The proatherosclerotic effect of PD-1 and CTLA4 inhibition seems to be, at least in part, mediated by the increase in INFγ and TNFα secretion, which are known mediators promoting coronary thrombosis [217]. As new immunomodulatory targets are discovered, it is interesting to see whether it can be possible to activate the immune response without increasing atherosclerotic risk [205].
Events of coronary spasm following ICI administration have been reported [218,219], but the full extent of this phenomenon warrants further investigation.
6. Emerging Treatment of Cardiac IRAEs
To date, the hallmark of IRAE management is based on steroid therapy and escalation to other immunosuppressive regiments in non-responders [7,153]. Immune suppression using the inosine-5′-monophosphate dehydrogenase (IMPDH) inhibitor, mycophenolate mofetil (MMF), results primarily in B- and T-cell inhibition [235,236]. Calcineurin inhibitors, such as tacrolimus, exert immunosuppressive properties through inhibiting T-cell activity [167,237,238,239]. Lymphopenia may be induced by antithymocyte globulin (ATG) [167,237,238], resulting in the cessation of autoimmunity. Other strategies include the use of intravenous immunoglobulin (IVIG) [191,192,193,240,241,242,243], and plasmapheresis [194,195,240,244,245,246] to incapacitate or remove ICIs (which have a long half-life of up to 27 days in some cases [201] can be employed.
Recent advances in the understanding of IRAE mechanisms have allowed the employment of targeted therapies. These include CTLA4-Ig (abatacept) to reimpose inhibition of activated T cells in myocarditis, as shown in mouse models [173] and case reports in humans [173,247]. Anti-CD52 (alemtuzumab), commonly used in CLL and multiple myeloma, has shown T-cell depletion capabilities and has been used in a case report with encouraging results [248]. Anti-TNFα (infliximab) has also shown promising results [153,166,240,249], although its utility in heart failure patients may be limited. It is worth noting that any treatment resulting in global quenching of the immune response also has the potential to allow tumor escape from immunosurveillance and disease progression. A better understanding of both the stimulatory and inhibitory molecules implicated in intracellular signaling pathways may allow better control of adverse events while allowing the tenacious antitumoral activity.
This entry is adapted from the peer-reviewed paper 10.3390/vaccines10040540
This entry is offline, you can click here to edit this entry!