Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kazumasa Wakamatsu | -- | 2196 | 2022-04-14 05:09:57 | | | |
| 2 | Catherine Yang | Meta information modification | 2196 | 2022-04-14 05:20:49 | | | | |
| 3 | Kazumasa Wakamatsu | Meta information modification | 2196 | 2022-04-15 03:51:06 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wakamatsu, K.; Nagatsu, T.; Nakashima, A.; Watanabe, H.; Ito, S. Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase. Encyclopedia. Available online: https://encyclopedia.pub/entry/21744 (accessed on 24 July 2026).
Wakamatsu K, Nagatsu T, Nakashima A, Watanabe H, Ito S. Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase. Encyclopedia. Available at: https://encyclopedia.pub/entry/21744. Accessed July 24, 2026.
Wakamatsu, Kazumasa, Toshiharu Nagatsu, Akira Nakashima, Hirohisa Watanabe, Shosuke Ito. "Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase" Encyclopedia, https://encyclopedia.pub/entry/21744 (accessed July 24, 2026).
Wakamatsu, K., Nagatsu, T., Nakashima, A., Watanabe, H., & Ito, S. (2022, April 14). Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase. In Encyclopedia. https://encyclopedia.pub/entry/21744
Wakamatsu, Kazumasa, et al. "Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase." Encyclopedia. Web. 14 April, 2022.
Copy Citation
Parkinson’s disease (PD) is an aging-related disease and the second most common neurodegenerative disease after Alzheimer’s disease. The main symptoms of PD are movement disorders accompanied with deficiency of neurotransmitter dopamine (DA) in the striatum due to cell death of the nigrostriatal DA neurons. Two main histopathological hallmarks exist in PD: cytosolic inclusion bodies termed Lewy bodies that mainly consist of α-synuclein protein, the oligomers of which produced by misfolding are regarded to be neurotoxic, causing DA cell death; and black pigments termed neuromelanin (NM) that are contained in DA neurons and markedly decrease in PD.
neuromelanin
norepinephrine
Parkinson’s disease
dopamine
substantia nigra
tyrosinase
tyrosine hydroxylase
melanin
locus coeruleus
1. Neuromelanin (NM) in Parkinson’s Disease
Parkinson’s disease (PD) is a human-specific, progressive, aging-related disease, and the second most common neurodegenerative disease after Alzheimer’s disease [1]. In 1817, James Parkinson in London published “An Essay on the Shaking Palsy”, the first comprehensive clinical description of a disorder later named Parkinson’s disease. The main symptoms of PD are motor symptoms, such as tremor, bradykinesia, rigidity, and postural instability, as well as non-motor symptoms including anosmia, constipation, insomnia, REM-sleep behavioral disorders (RBD), anxiety, depression, fatigue, and cognitive impairment [1]. Most PD is sporadic without a familial history (sPD). Only 5–15 percent of cases are familial PD (fPD) [2][3]. The pathophysiology of PD was investigated by biochemical analysis of post-mortem PD brains during the middle of 20th century [4][5][6][7]. Although the pathophysiology of PD still remains unknown, sPD is thought to be caused by combined effects of environmental and genetic factors. The main symptoms of PD, which is a movement disorder, are known to be caused by a decrease in neurotransmitter dopamine (DA) in the striatum in the basal ganglia due to neurodegeneration of nigrostriatal DA neurons, and supplementation of DA by the direct precursor L-3,4-dihydroxyphenylalanine (L-DOPA) is still the gold standard of pharmacotherapy of PD after five decades since 1970s [1][7][8]. L-DOPA treatment is highly effective for alleviating many core symptoms of PD, but it does not prevent the progression of neurodegeneration and later results in a decrease in efficacy and various side effects such as dyskinesia [7][8].
The discovery of the causative or susceptibility genes of various fPD, since the end of 20th century, has greatly promoted the elucidation of molecular mechanism of sPD [3]; fPD is termed in the order of discovery of the gene loci such as PARK1 (α-synuclein, SNCA [9][10]) and PARK2 (parkin, PRKN [3][11][12]). More than 20 PARKs have been reported. The abbreviation PARK is derived from the name PARKinson. Mutations in some genes in fPD are considered to be causative and also related to susceptibility loci in sPD, for example, α-synuclein gene (SNCA and PARK1) [9][10], parkin (PARK2) [3][11], PTEN-induced putative kinase 1 (PINK1 and PARK6) [13][14], and leucine-rich repeat kinase 2 (LRRK2 and PARK8) [15][16][17][18].
There are two main histopathological hallmarks in PD in the degenerating nigrostriatal DA neurons, i.e., Lewy bodies and reduction of neuromelanin (NM) in substantia nigra (SN) (Figure 1): (1) Friedrich Heinrich described cytosolic inclusion bodies termed Lewy bodies in 1912 [19]. Lewy bodies contain α-synuclein protein as the main protein component, and the fibrillar oligomers of α-synuclein protein produced by misfolding are presumed to be neurotoxic and to cause DA cell death [20]. Mutation of the α-synuclein gene (SNCA) was found, in 1997, to cause a dominant fPD (PARK1) in which degenerating dopamine neurons contain both Lewy bodies containing α-synuclein and black pigment NM [9][10]. For these reasons, the α-synuclein protein has been extensively examined in relation to DA neuron death in sPD. However, a remaining question is that Lewy bodies are observed in dominant fPD such as PARK1 (SNCA), but not in recessive fPD such as PARK2 (PARKIN). (2) A black pigment NM, which is observed in the human SN, gradually increases during normal aging in healthy subjects [21]. NM is rich in the brain of humans, but the presence is also reported in the brain of monkeys, mice, rats, dogs, and horses [22][23]. Konstantin Tretiakoff [24], in 1919, reported that NM markedly decreased in the SN of PD brains. A decrease in NM in some nigrostriatal DA neurons in the SN pars compacta (SNpc), visible with the naked eye, are the main histopathological sign of PD. Different from Lewy bodies, NM is observed in sPD, dominant fPD, and recessive fPD. NM is also contained in norepinephrine (NE) neurons in the human locus coeruleus (LC), where NE neurons also degenerate in PD. In contrast to α-synuclein protein in Lewy bodies that has received great attention, the biosynthesis and pathophysiology of NM in PD remain less known. One reason is that elucidation of chemical structures of NM has been difficult owing to the small contents only in the postmortem human brains. However, the chemical properties and the biosynthesis pathway of NM has been elucidated in the last two decades based on the development of chemical micro-analysis of NM isolated from the SN of post-mortem human brains [25][26][27], and the pathophysiology of NM has also been gradually elucidated.
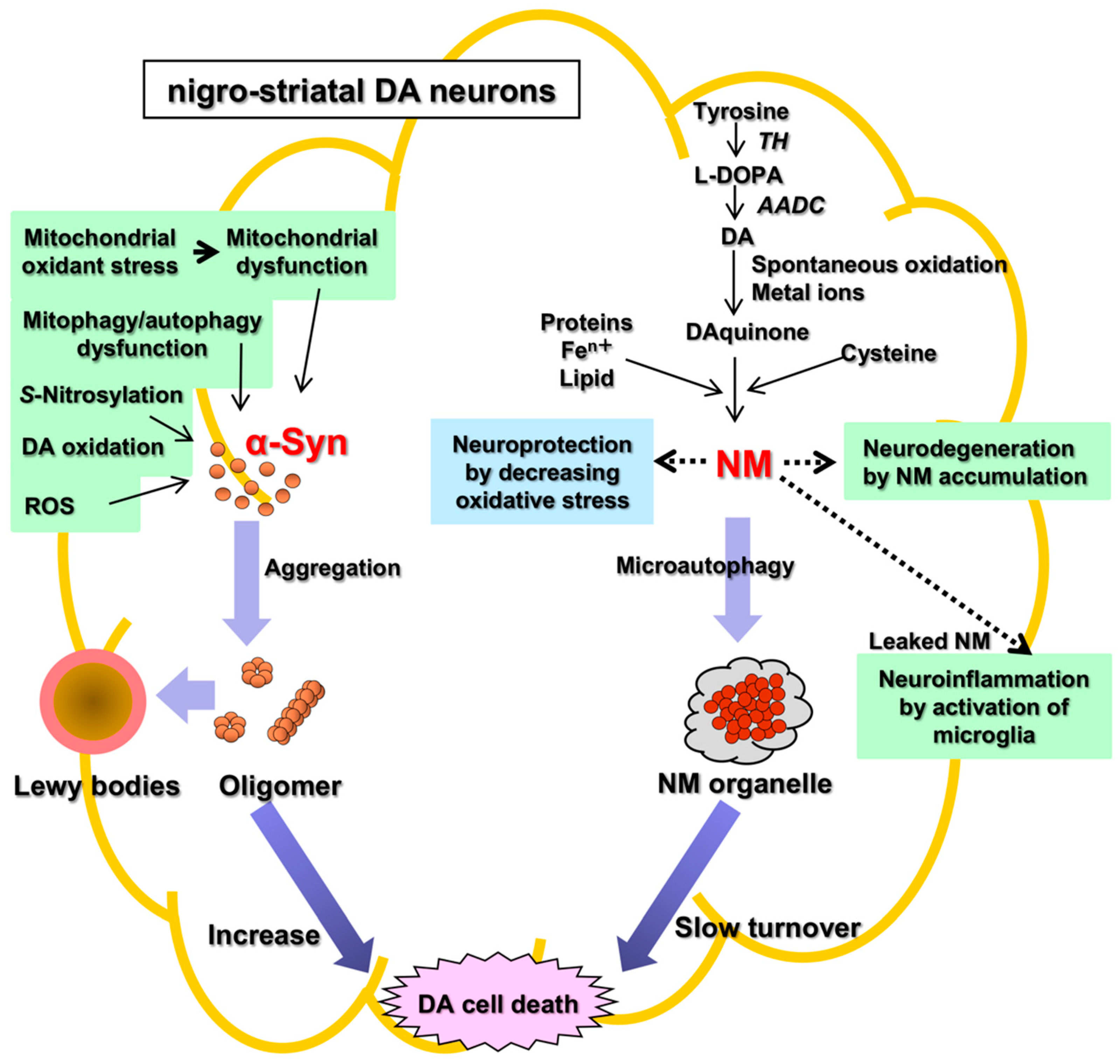
Figure 1. Two histopathological hallmarks in PD in the nigrostriatal DA. Fibrillar oligomers of α-Syn produced by misfolding are presumed to be neurotoxic and to cause DA cell death. Neuromelanin (NM) is also related to neurodegeneration and DA cell death, because NM attenuates the oxidative stress for neuroprotection. α-Syn, α-synuclein; NM, neuromelanin; TH, tyrosine hydroxylase; AADC, aromatic amino acid decarboxylase; ROS, reactive oxygen species.
2. Neuromelanin (NM): The Cause of Parkinson’s Disease?
The pathophysiology of PD remains unknown. There are two hypotheses of cell death of DA neurons based on two histopathological hallmarks in PD, i.e., the α-synuclein hypothesis (an α-synucleinopathy) and the NM hypothesis (Figure 1). The α-synuclein hypothesis on the possible molecular mechanism of neuronal death of DA neurons in sPD may be summarized as follows: Mitochondrial oxidant stress by various exogeneous or endogenous factors may produce mitochondrial dysfunction, especially complex I deficiencies [28][29][30][31][32][33], oxidation of DA in cytoplasm [34][35][36], and formation of oxidized DA accumulation, especially toxic 3,4-dihydroxyphenylacetaldehyde (DOPAL), formation of toxic ROS, accumulation of cytotoxic fibrillar aggregates of α-synuclein oligomers, mitophagy/autophagy dysfunction, and neuroinflammation [37][38][39][40][41][42][43][44]. DOPAL is thought to accumulate in PD due to the low aldehyde dehydrogenase activity that oxidizes DOPAL to DOPAC in the SN in PD [45] and DOPAL generates potential reactive intermediates as causative agents for its neurotoxicity [46][47].
Since the 1990s, it was found that Lewy bodies mainly consisted of α-synuclein protein, and that the fibrillar oligomers produced by misfolding of the protein were neurotoxic and may be related to the cause of DA cell death [42][48][49]. In 1997, mutation of the α-synuclein gene (SNCA) was found to cause a familial PD (PARK1) [9][10]. Prion-like properties of α-synuclein were proposed by Braak (Braak hypothesis); α-synuclein produced in the intestine or olfactory bulb might spread via the vagus nerve or olfactory pathway to the midbrain and basal ganglia by cell-to-cell transfer [50][51][52]. α-Synuclein aggregates may spread from neuron to neuron, apparently transmitting the disease process through the brain. However, precisely how α-synuclein aggregates build-up and spread in this way is still unknown. Another question is that α-synuclein is not specific to PD, and also found in Lewy body disease (LBD) and multiple system atrophy (MSA) [53]. Aggregates of α-synuclein in distinct synucleinopathies, PD and MSA, have been proposed to represent different conformational strains of α-synuclein [54]. Even with these questions about the α-synuclein hypothesis, α-synuclein has been extensively examined in relation to DA neuron death in PD. The p62 protein normally assists in autophagy, a waste-management system that helps cells get rid of potentially harmful protein aggregates. In cell and animal models of PD, p62 is S-nitrosylated at abnormally high levels in affected neurons. This alteration of p62 inhibits autophagy, causing a build-up of α-synuclein aggregates, which in turn, leads to the secretion of segregates by affected neurons, and some of these aggregates are taken up by nearby neurons [55]. There are many references to support the cytotoxic effects of α-synuclein in vitro, especially in cell culture systems [10][20][42]. A downsized and optimized intracellular library-derived peptide prevents α-synuclein primary nucleation and toxicity without impacting upon lipid binding [56]. An animal model of PD with prodromal symptoms as in human PD has been reported [57]. The α-synuclein gene, SNCA, is a risk gene for sPD. A bacterial artificial chromosome transgenic mouse harboring SNCA and its gene expression regulating region in order to maintain the native expression pattern of α-synuclein showed prodromal symptoms in human PD such as RBD and anosmia without motor symptoms [57][58]. This mouse model is similar to human sPD and shows that α-synuclein alone can cause PD [57].
In the NM hypothesis, NM alone is related to DA neuron death. This entry focuses on two independent ways of PD pathology, α-synuclein and NM. While both pathways may indeed lead to dopaminergic cell death, a decisive link between them is proposed “iron”, as pointed out recently by Riederer et al. [53][59][60]. The pathophysiology of NM decrease in the SN of DA neurons as a hallmark of PD remains unknown, especially in its relation to DA neuron death. In parallel with the elucidation of the chemistry and biosynthesis of NM in the DA neurons in the SN in PD, the physiological and pathological roles on NM have been studied since 2000s. NM in the SN increases gradually during aging in healthy subjects [21]. In contrast, NM decreases in PD. In PD, DA neurons containing NM in the human SN might preferentially degenerate, in parallel with the marked reduction in NM in the SN [61]. This fact, although controversial, suggests that NM is related to neurodegeneration and DA neuron death.
Alternatively, NM in DA neurons has generally been regarded as acting for neuroprotection, since NM inactivates toxic free radical species via its ability to chelate transition metals, especially iron. Iron also accumulates in DA neurons [59][62][63][64]. Iron is bound to NM in the ferrous (II) iron form, a redox-active form that is involved in a Fenton-like reaction to produce toxic free radical species. NM also eliminates various toxic substances in cytoplasm. Thus, NM may act for neuroprotection also in vivo. However, during the progress of PD, the release of toxic substances bound to NM owing to intracellular NM degradation may result in activation of microglia to release cytotoxic cytokines that produce neuroinflammation and neurodegeneration [65][66]. PD occurs spontaneously only in humans. To produce the PD phenotype in various models of PD in mammals such as in mice and rats that nearly lack NM in the brain, it is necessary to trigger the DA neurodegeneration by some toxic chemicals such as 1-phenyl-4-methyl-1,2,3,6-tetrahydropyridine (MPTP) that inhibits the mitochondrial complex I [44]. Vila’s group reported that NM accumulation in DA neurons during aging over a threshold causes DA neuron death and PD phenotype [67][68][69]. They created a rat model of human PD by overexpression of human NM in the right SNpc by stereotaxic injection of an adeno-associated viral (AAV) vector expressing human tyrosinase [67]. The rats showed age-dependent production of human-like NM within nigral DA neurons, up to levels in elderly humans. Intracellular NM aggregation above a specific threshold is associated with an age-dependent PD phenotype, including hypokinesia. Enhancing lysosomal proteostasis reduces intracellular NM and prevents neurodegeneration in tyrosinase-overexpressing rats. Intracellular NM levels may set the threshold for the initiation of PD. Furthermore, extracellular NM leaked from dead NM-containing DA neurons may activate microglia to produce neuroinflammation and to further promote DA cell death [70].
NM is a hot candidate to trigger PD and/or to lead to progressed degeneration of PD, however, there are unsolved problems arising from the treatment of PD with levodopa: (1) Post-mortem data have not shown an increase of NM in surviving NM containing dopaminergic neurons of the SN after levodopa long term treatment and (2) long term levodopa treatment has not demonstrated a significant increase in the progression of PD. The NM theory well fit to the phenotypes of human sPD. In addition, there is much evidence on the cytotoxicity of α-synuclein [10][19][71].
Recently, the role of NM in inducing α-synuclein expression and aggregation has been suggested as a mechanism for this pigment to modulate neuronal vulnerability in PD [72]. α-Synuclein reacts with tyrosinase, and the chemical modifications on the tyrosinase-treated α-synuclein strongly influence its aggregation properties and increase the toxicity, and α-synuclein may influence synthesis of NM [73][74]. Iron redox chemistry promotes the aggregation of α-synuclein, and protein-metal complex aggregates are directly involved in ROS production, exacerbating the oxidative damage [75]. Furthermore, DA neurons easily express MHC-I, and induction of MHC-I is promoted by activation of microglia either by α-synuclein or by NM, as well as by interferon gamma or high cytosolic DA and oxidative stress [76]. The activated microglia in PD brains express major histocompatibility complex class II (MHC-II) molecules. The number of MHC-II positive microglia in the SN and putamen increase as the neuronal degeneration of the SN proceeds [77].
An evolution theory has been proposed to explain human-specific PD based on the greater development of human cerebral cortex than that of basal ganglia [78][79][80]. Clinically, PD is a systemic disease, and it is difficult to explain the degenerative processes, especially in the autonomic nervous system, exclusively by NM theory, although there is accumulating evidence that the pathogenesis of PD is complex and involves energy metabolism disorders, oxidative stress, proteasomal abnormalities, α-synuclein accumulation, alterations of gut microbiota metabolites, and neuroinflammation [81][82]. In this context, the evolutional point of view on the NM system and α-synuclein system is also of interest.
References
- Cacabelos, R. Parkinson’s disease: From pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551.
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. Acad. Neurol. 2020, 27, 27–42.
- Guadagnolo, D.; Piane, M.; Torrisi, M.R.; Pizzuti, A.; Petrucci, S. Genotype-phenotype correlations in monogenic Parkinson disease: A review on clinical and molecular findings. Front. Neurol. 2021, 12, 648588.
- Lloyd, K.G.; Davidson, L.; Hornykiewicz, O. The neurochemistry of Parkinson’s disease: Effect of L-DOPA therapy. J. Pharmacol. Exp. Ther. 1975, 153, 453–464.
- Nagatsu, T.; Kato, T.; Numata (Sudo), Y.; Ikuta, K.; Sano, M.; Nagatsu, I.; Kondo, Y.; Inagaki, S.; Iizuka, R.; Hori, A.; et al. Phenylethanolamine N-methyltransferase and other enzymes of catecholamine metabolism in human brain. Clin. Chim. Acta 1977, 75, 221–232.
- Nagatsu, T.; Sawada, M. Biochemistry of postmortem brains in Parkinson’s disease: Historical overview and future prospects. J. Neural Transm. Suppl. 2007, 72, 113–120.
- Fahn, S. The medical treatment of Parkinson disease from James Parkinson to George Cotzias. Mov. Disord. 2015, 30, 4–18.
- Nagatsu, T.; Sawada, M. L-DOPA therapy for Parkinson’s disease: Past, present, and future. Parkinsonism. Relat. Disord. 2009, 15, S3–S8.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boye, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257.
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokoti, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608.
- Gundogdu, M.; Tadayon, R.; Salzano, G.; Shaw, G.S.; Walden, H. A mechanistic review of Parkin activation. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129894.
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273.
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signaling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189.
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRKK2 links to genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661.
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRKK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107.
- Watanabe, R.; Buschauer, R.; Böhning, J.; Audagnotto, M.; Lasker, K.; Lu, T.-W.; Boassa, D.; Taylor, S.; Villa, E. The In Situ structure of Parkinson’s disease-linked LRRK2. Cell 2020, 182, 1508–1518.
- Erb, M.L.; Moore, D.J. LRRK2 and endolysosomal system in Parkinson’s disease. J. Parkinson’s Dis. 2020, 10, 1271–1291.
- Holdorff, B. Friedrich Heinrich Lewy (1885–1950) and his work. J. Hist. Neurosci. 2002, 11, 19–28.
- Mehra, S.; Sahay, S.; Maji, S.K. α-synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908.
- Zecca, L.; Fariello, R.; Riederer, P.; Sulzer, D.; Gatti, A.; Tampellini, D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. FEBS Lett. 2002, 510, 216–220.
- Barden, H. The histochemical distribution and localization of copper, iron, neuromelanin and lysosomal enzyme activity in the brain of aging rhesus monkey and the dog. J. Neuropathol. Exp. Neurol. 1971, 30, 650–657.
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia nigra: An update. Neurotox. Res. 2014, 25, 13–23.
- Holdorff, B. Centenary of Tretiakoff’s thesis on the morphology of Parkinson’s disease, evolved on the grounds of encephalitis lethargica pathology. J. Hist. Neurosci. 2019, 28, 387–398.
- Wakamatsu, K.; Fujikawa, K.; Zucca, F.A.; Zecca, L.; Ito, S. The structure of neuromelanin as studied by chemical degradative methods. J. Neurochem. 2003, 86, 1015–1023.
- Wakamatsu, K.; Ohtara, K.; Ito, S. Chemical analysis of late stages of pheomelanogenesis: Conversion of dihydrobenzothiazine to a benzothiazole structure. Pigment Cell Melanoma Res. 2009, 22, 474–486.
- Wakamatsu, K.; Murase, T.; Zucca, F.A.; Zecca, L.; Ito, S. Biosynthetic pathway to neuromelanin and its aging process. Pigment Cell Melanoma Res. 2012, 25, 792–803.
- Mizuno, Y.; Ohta, S.; Tanaka, S.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1989, 163, 1450–1455.
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269.
- Reichmann, H.; Riederer, P. Biochemische Analyse der Atmungskettenkomplex verschiedener Hirnregionen von Patienten mit M. Parkinson. In Proceedings of the Symposium zu Morbus Parkinson und andere Basalganglienerkrankungen, Bad Kissingen, Germany, 23–25 April 1989; Ministerium für Forschung und Technologie (BMBF): Bad Kissingen, Germany, 1989; p. 44.
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gaol, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656.
- Hattori, N.; Saiki, S.; Imai, Y. Regulation by mitophagy. Int. J. Biochem. Cell Biol. 2014, 53, 147–150.
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial dysfunction and mitophagy in Parkinson’s disease; from mechanism to therapy. Trends Biochem. Sci. 2021, 46, 329–343.
- Monzani, E.; Nicolis, S.; Dell’Acqua, S.; Capucciati, A.; Bacchella, C.; Zucca, F.A.; Mosharov, E.V.; Sulzer, D.; Zecca, L.; Casella, L. Dopamine, oxidative stress and protein-quinone modifications in Parkinson’s and other neurodegenerative diseases. Angew. Chem. Int. Ed. Engl. 2019, 58, 6512–6527.
- Sian-Hulsmann, J.; Riederer, P. The nigral coup in Parkinson’s disease by α-synuclein and its associated rebels. Cells 2021, 10, 598.
- Wright, R. Mitochondrial dysfunction and Parkinson’s disease. Nat. Neurosci. 2021, 599, 650–656.
- Cook, C.; Petrucelli, L. A Critical evaluation of the ubiquitin-proteasome system in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 664–675.
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544.
- Reeve, A.K.; Ludtman, M.H.R.; Angelova, P.R.; Simcox, E.M.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Turnbull, D.M.; Abramov, A.Y. Aggregated α-synuclein and complex I deficiency: Explorations of their relationship in differentiated neurons. Cell Death Dis. 2015, 6, e1820.
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nature Neurosci. 2017, 20, 1560–1568.
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wang, Y.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261.
- Burré, J.; Sharma, M.; Südhof, T.C. Cell biology and pathophysiology of α-synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091.
- Guiney, S.F.; Adlard, P.A.; Lei, P.; Mawal, C.H.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Fibrillar α-synuclein toxicity depends on functional lysosomes. J. Biol. Chem. 2020, 295, 17497–17513.
- Dionísio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative stress and regulated cell death in Parkinson’s disease. Aging Res. Rev. 2021, 67, 101263.
- Grünblatt, E.; Riederer, P. Aldehyde dehydrogenase (ALDH) in Alzheimer’s and Parkinson’s disease. J. Neural Transm. 2016, 123, 83–90.
- Goldsein, D.S. The catecholaldehyde hypothesis: Where MAO fits in. J. Neural Transm. 2020, 127, 169–177.
- Ito, S.; Tanaka, H.; Ojika, M.; Wakamatsu, K.; Sugumaran, M. Oxidative transformations of 3,4-dihydroxyphenylacetaldehyde generate potential reactive intermediates as causative agents for its neurotoxicity. Int. J. Mol. Sci. 2021, 22, 11751.
- Sahay, S.; Ghosh, D.; Singh, P.K.; Maji, S.K. Alteration of structure and aggregation of α-synuclein by familial Parkinson’s disease associated mutations. Curr. Protein Pept. Sci. 2017, 18, 656–676.
- Double, K.L.; Ben-Shachar, D.; Youdim, M.B.H.; Zecca, L.; Riederer, P.; Gerlach, M. Influence of neuromelanin on oxidative pathways within the human substantia nigra. Neurotoxicol. Teratol. 2002, 24, 621–628.
- McCann, H.; Cartwright, H.; Halliday, G.M. Neuropathology of α-synuclein progression and Braak hypothesis. Mov. Disord. 2016, 31, 152–160.
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.-M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.-J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1-/- mice. Nature 2019, 571, 569.
- Jia, L.; Liu, Y.; Wang, W.; Wang, Y.; Liu, H.; Liu, F.; Chen, R.; Dawson, V.L.; Dawson, T.M.; Lu, F.; et al. Molecular mediation of prion-like α-synuclein fibrillation from toxic PFFs to nontoxic species. ACS App. Biol. Mater. 2020, 3, 6096–6102.
- Riederer, P.; Berg, D.; Casadei, N.; Cheng, F.; Classen, J.; Dresel, C.; Jost, W.; Krüger, R.; Müller, T.; Reichmann, H.; et al. α-Synuclein in Parkinson’s disease: Causal or bystanders? J. Neural. Transm. 2019, 126, 815–840.
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277.
- Oh, C.-K.; Dolatabadi, N.; Cieplak, P.; Diaz-Meco, M.T.; Moscat, J.; Nolan, J.P.; Nakashima, T.; Lipton, S.A. S-Nitrosylation of p62 inhibits autophagic flux to promote α-synuclein secretion and spread in Parkinson’s disease and Lewy body dementia (LBD). J. Neurosci. 2022, 42, 3011–3024.
- Meade, R.M.; Watt, K.J.C.; Williams, R.J.; Mason, J.M. A downsized and optimized intracellular library-derived peptide prevents alpha-synuclein primary nucleation and toxicity without impacting upon lipid binding. J. Mol. Biol. 2021, 433, 167323.
- Taguchi, T.; Ikuno, M.; Hondo, M.; Parajuli, L.K.; Taguchi, K.; Ueda, J.; Sawamura, M.; Okuda, S.; Nakanishi, E.; Hara, J.; et al. α-Synuclein BAC transgenic mice exhibit RBD-like behaviour and hyposmia: A prodromal Parkinson’s disease model. Brain 2020, 143, 249–265.
- Blesa, J.; Foffani, G.; Dehay, B.; Bezard, E.; Obeso, J.A. Motor and non-motor circuits disturbances in early Parkinson disease: Which happen first? Nat. Rev. Neurosci. 2022, 23, 115–128.
- Götz, M.E.; Doulble, K.; Gerlach, M.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2004, 1012, 193–208.
- Riederer, P.; Monoranu, C.; Strobel, S.; Iordache, T.; Sian-Hülsmann, J. Iron as the concert master in the pathogenic orchestra playing in sporadic Parkinson’s disease. J. Neural. Transm. 2021, 128, 1577–1598.
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988, 334, 345–348.
- Youdim, M.B.; Ben-Shachar, D.; Riederer, P. Iron in brain function and dysfunction with emphasis on Parkinson’s disease. Eur. Neurol. 1991, 31, 34–40.
- Gerlach, M.; Double, K.L.; Ben-Shachar, D.; Zecca, L.; Youdim, M.B.H.; Riederer, P. Neuromelanin and its interaction with iron as a potential risk factor for dopaminergic neurodegeneration underlying Parkinson’s disease. Neurotox. Res. 2003, 5, 35–44.
- Mochizuki, H.; Choong, C.-J.; Baba, K. Parkinson’s disease and iron. J. Neural Transm. 2020, 127, 181–187.
- Zecca, L.; Bellei, C.; Costi, P.; Albertini, A.; Monzani, E.; Casella, L.; Gallorini, M.; Bergamaschi, L.; Moscatelli, A.; Turro, N.J.; et al. New melanic pigments in the human brain that accumulate in aging and block environmental toxic metals. Proc. Natl. Acad. Sci. USA 2008, 105, 17567–17572.
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interaction of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119.
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973.
- Vila, M. Neuromelanin, aging, and neuronal vulnerability in Parkinson’s disease. Mov. Disord. 2019, 34, 1440–1451.
- Vila, M.; Laguna, A.; Carballo-Carbajal, I. Intracellular crowding by age-dependent neuromelanin accumulation disrupts neuronal proteostasis and triggers Parkinson disease pathology. Autophagy 2019, 15, 2028–2030.
- Zucca, F.A.; Giaveri, G.; Gallorini, M.; Albertini, A.; Toscani, M.; Pezzoli, G.; Lucius, R.; Wilms, H.; Sulzer, D.; Ito, S.; et al. The neuromelanin of human substantia nigra: Physiological and pathogenic aspects. Pigment Cell Res. 2004, 17, 610–617.
- Lin, K.-S.; Lin, K.-L.; Chen, S.-D.; Liou, C.-W.; Chuang, Y.-C.; Lin, H.-Y.; Lin, T.-K. The overcrowded crossroads: Mitochondria, alpha-synuclein, and the endo-lysosomal system interaction. Int. J. Mol. Sci. 2019, 20, 5312.
- Xu, S.; Chan, P. Interaction between neuromelanin and alpha-synuclein in Parkinson’s disease. Biomolecules 2015, 5, 1122–1142.
- Tessari, I.; Bisaglia, M.; Valle, F.; Samori, B.; Bergantino, E.; Mammi, S.; Bubacco, L. The reaction of α-synuclein with tyrosinase: Possible implication for Parkinson’s disease. J. Biol. Chem. 2008, 283, 16808–16817.
- Pan, T.; Zhu, J.; Hwu, W.-J.; Jankovic, J. The role of alpha-synuclein in melanin synthesis in melanoma and dopaminergic neuronal cells. PLoS ONE 2012, 7, e45183.
- Moreno-García, A.; Kun, A.; Calero, M.; Calero, O. The neuromelanin paradox and its dual role in oxidative stress and neurodegeneration. Antioxidants 2021, 10, 124.
- Cebrian, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsatell, J.P.; et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 2014, 5, 3633.
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526.
- Diederich, N.J.; Surmeier, D.J.; Uchihara, T.; Grillner, S.; Goetz, C.G. Parkinson’s disease: Is it a consequence of human brain evolution? Mov. Disord. 2019, 34, 453–459.
- Foffani, G.; Obeso, J.A. A cortical pathogenic theory of Parkinson’s disease. Neuron 2018, 99, 1116–1128.
- Diederich, N.J.; Uchihara, T.; Grillner, S.; Goetz, C.G. The evolution-driven signature of Parkinson’s disease. Trends Neurosci. 2020, 43, 475–492.
- Liang, Y.; Cui, L.; Gao, J.; Zhu, M.; Zhang, Y.; Zhang, H.L. Gut microbial metabolites in Parkinson’s disease: Implications of mitochondrial dysfunction in the pathogenesis and treatment. Mol. Neurobiol. 2021, 58, 3745–3758.
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 1–17.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Entry Collection:
Neurodegeneration
Revisions:
3 times
(View History)
Update Date:
15 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No