Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 3 by Kazumasa Wakamatsu.
Parkinson’s disease (PD) is an aging-related disease and the second most common neurodegenerative disease after Alzheimer’s disease. The main symptoms of PD are movement disorders accompanied with deficiency of neurotransmitter dopamine (DA) in the striatum due to cell death of the nigrostriatal DA neurons. Two main histopathological hallmarks exist in PD: cytosolic inclusion bodies termed Lewy bodies that mainly consist of α-synuclein protein, the oligomers of which produced by misfolding are regarded to be neurotoxic, causing DA cell death; and black pigments termed neuromelanin (NM) that are contained in DA neurons and markedly decrease in PD.
- neuromelanin
- norepinephrine
- Parkinson’s disease
- dopamine
- substantia nigra
- tyrosinase
- tyrosine hydroxylase
- melanin
- locus coeruleus
1. Neuromelanin (NM) in Parkinson’s Disease
Parkinson’s disease (PD) is a human-specific, progressive, aging-related disease, and the second most common neurodegenerative disease after Alzheimer’s disease [1]. In 1817, James Parkinson in London published “An Essay on the Shaking Palsy”, the first comprehensive clinical description of a disorder later named Parkinson’s disease. The main symptoms of PD are motor symptoms, such as tremor, bradykinesia, rigidity, and postural instability, as well as non-motor symptoms including anosmia, constipation, insomnia, REM-sleep behavioral disorders (RBD), anxiety, depression, fatigue, and cognitive impairment [1]. Most PD is sporadic without a familial history (sPD). Only 5–15 percent of cases are familial PD (fPD) [2][3]. The pathophysiology of PD was investigated by biochemical analysis of post-mortem PD brains during the middle of 20th century [4][5][6][7]. Although the pathophysiology of PD still remains unknown, sPD is thought to be caused by combined effects of environmental and genetic factors. The main symptoms of PD, which is a movement disorder, are known to be caused by a decrease in neurotransmitter dopamine (DA) in the striatum in the basal ganglia due to neurodegeneration of nigrostriatal DA neurons, and supplementation of DA by the direct precursor L-3,4-dihydroxyphenylalanine (L-DOPA) is still the gold standard of pharmacotherapy of PD after five decades since 1970s [1][7][8]. L-DOPA treatment is highly effective for alleviating many core symptoms of PD, but it does not prevent the progression of neurodegeneration and later results in a decrease in efficacy and various side effects such as dyskinesia [7][8].
The discovery of the causative or susceptibility genes of various fPD, since the end of 20th century, has greatly promoted the elucidation of molecular mechanism of sPD [3]; fPD is termed in the order of discovery of the gene loci such as PARK1 (α-synuclein, SNCA [9][10]) and PARK2 (parkin, PRKN [3][11][12]). More than 20 PARKs have been reported. The abbreviation PARK is derived from the name PARKinson. Mutations in some genes in fPD are considered to be causative and also related to susceptibility loci in sPD, for example, α-synuclein gene (SNCA and PARK1) [9][10], parkin (PARK2) [3][11], PTEN-induced putative kinase 1 (PINK1 and PARK6) [13][14], and leucine-rich repeat kinase 2 (LRRK2 and PARK8) [15][16][17][18].
There are two main histopathological hallmarks in PD in the degenerating nigrostriatal DA neurons, i.e., Lewy bodies and reduction of neuromelanin (NM) in substantia nigra (SN) (Figure 1): (1) Friedrich Heinrich described cytosolic inclusion bodies termed Lewy bodies in 1912 [19]. Lewy bodies contain α-synuclein protein as the main protein component, and the fibrillar oligomers of α-synuclein protein produced by misfolding are presumed to be neurotoxic and to cause DA cell death [20]. Mutation of the α-synuclein gene (SNCA) was found, in 1997, to cause a dominant fPD (PARK1) in which degenerating dopamine neurons contain both Lewy bodies containing α-synuclein and black pigment NM [9][10]. For these reasons, the α-synuclein protein has been extensively examined in relation to DA neuron death in sPD. However, a remaining question is that Lewy bodies are observed in dominant fPD such as PARK1 (SNCA), but not in recessive fPD such as PARK2 (PARKIN). (2) A black pigment NM, which is observed in the human SN, gradually increases during normal aging in healthy subjects [21]. NM is rich in the brain of humans, but the presence is also reported in the brain of monkeys, mice, rats, dogs, and horses [22][23]. Konstantin Tretiakoff [24], in 1919, reported that NM markedly decreased in the SN of PD brains. A decrease in NM in some nigrostriatal DA neurons in the SN pars compacta (SNpc), visible with the naked eye, are the main histopathological sign of PD. Different from Lewy bodies, NM is observed in sPD, dominant fPD, and recessive fPD. NM is also contained in norepinephrine (NE) neurons in the human locus coeruleus (LC), where NE neurons also degenerate in PD. In contrast to α-synuclein protein in Lewy bodies that has received great attention, the biosynthesis and pathophysiology of NM in PD remain less known. One reason is that elucidation of chemical structures of NM has been difficult owing to the small contents only in the postmortem human brains. However, the chemical properties and the biosynthesis pathway of NM has been elucidated in the last two decades based on the development of chemical micro-analysis of NM isolated from the SN of post-mortem human brains [25][26][27], and the pathophysiology of NM has also been gradually elucidated.
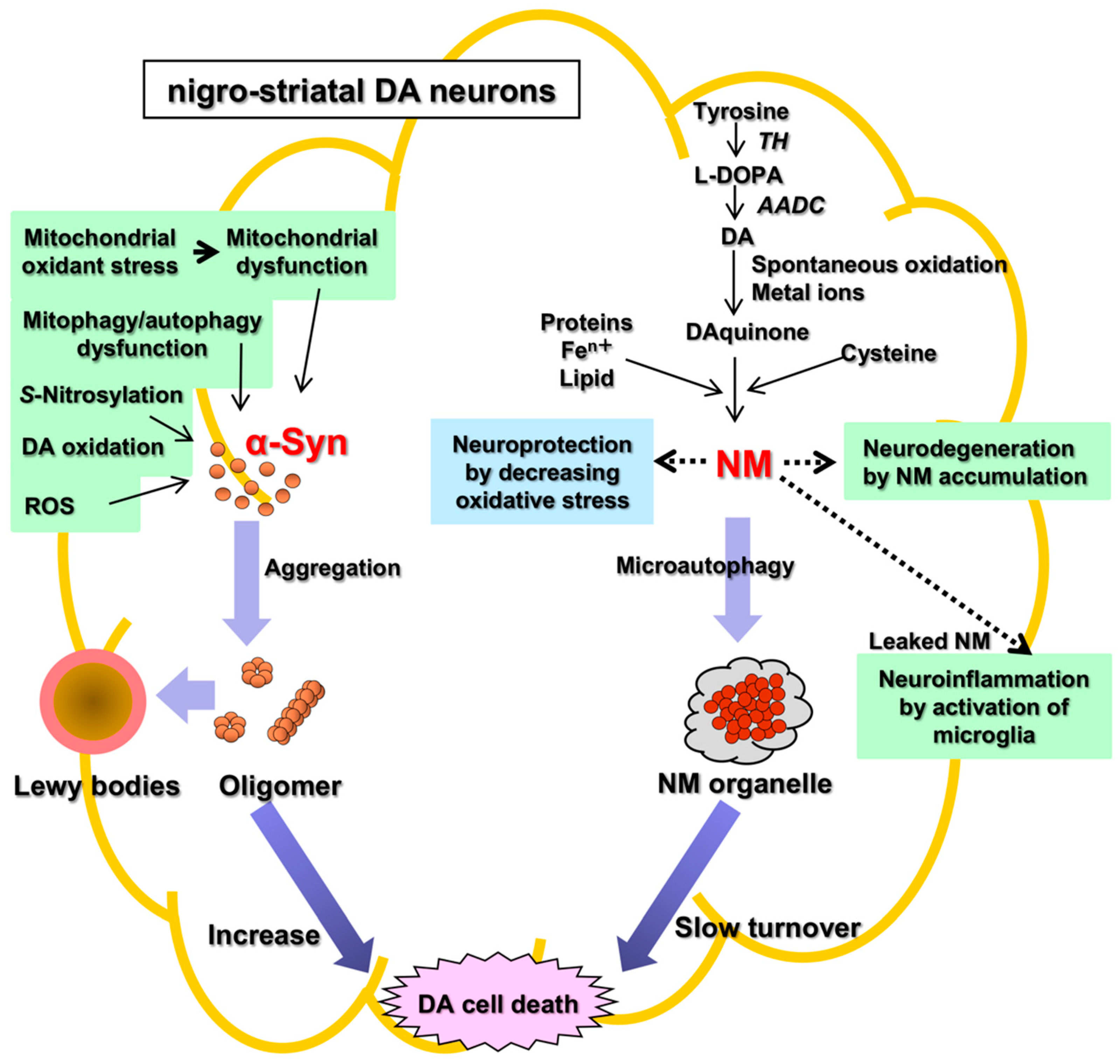
Figure 1. Two histopathological hallmarks in PD in the nigrostriatal DA. Fibrillar oligomers of α-Syn produced by misfolding are presumed to be neurotoxic and to cause DA cell death. Neuromelanin (NM) is also related to neurodegeneration and DA cell death, because NM attenuates the oxidative stress for neuroprotection. α-Syn, α-synuclein; NM, neuromelanin; TH, tyrosine hydroxylase; AADC, aromatic amino acid decarboxylase; ROS, reactive oxygen species.