 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrzej Kasperski | -- | 3451 | 2022-04-12 17:51:12 | | | |
| 2 | Jason Zhu | -11 word(s) | 3440 | 2022-04-13 03:52:36 | | | | |
| 3 | Jason Zhu | -61 word(s) | 3379 | 2022-05-06 05:52:27 | | | | |
| 4 | Jason Zhu | Meta information modification | 3379 | 2022-05-06 05:54:02 | | |
Video Upload Options
As a phenomenon, cancer is a disease related to multicellular evolution, i.e., cancer in general is understood to be a failure of the multicellular systems and is considered a reversal to unicellularity. Cancer cells are like unicellular organisms that benefit from ancestral-like traits. As a disease, cancer can be interpreted as (a) a destruction of cooperative behaviors underlying multicellular evolution, (b) a disruption of molecular networks established during the emergence of multicellularity or (c) an atavistic state resulting from reactivation of primitive programs typical of the earliest unicellular species. From this point of view and in accordance with the layered model of evolution of cellular functionalities, cancer transformation can occur as a result of huge disturbances or the destruction of functionalities that are located in the multicellular layer.
1. Introduction
2. Cancer Transformation as a Loss of Control over Atavistic Functionalities
3. Vertical and Horizontal Cancer Development
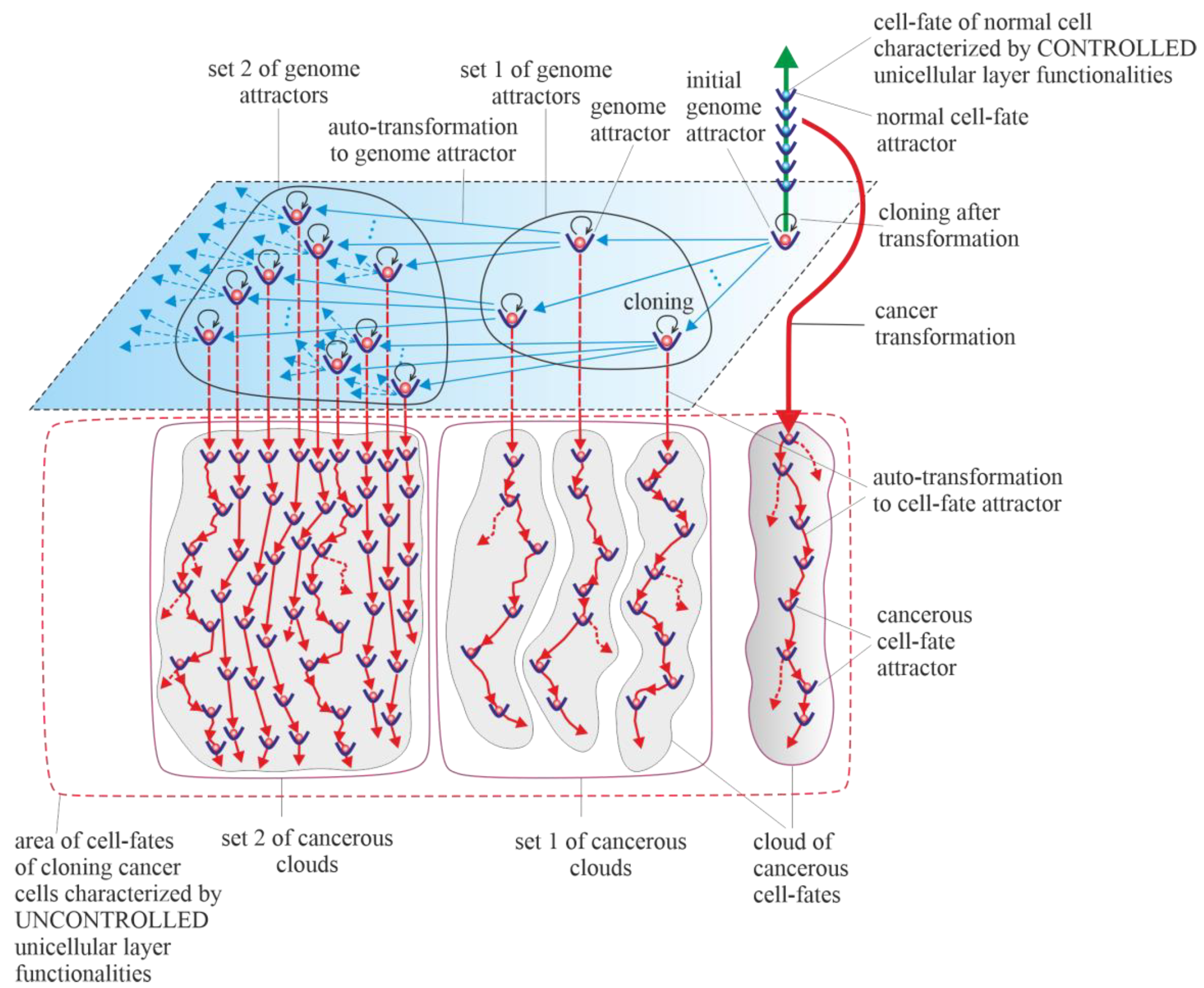
4. Vertical Cancer Development
5. Horizontal Cancer Development
6. Purely Vertical Cancer Development
7. Cancers without Mutation
8. Cancer Development as a Learning Process
References
- Heng, J.; Heng, H.H. Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”. Genes 2022, 13, 101.
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of cancer: Survival at the brink. Semin. Cancer Biol. 2020; in press.
- Heng, H.H. Debating Cancer: The Paradox in Cancer Research; World Scientific Publishing Co.: Singapore, 2015; ISBN 978-981-4520-84-3.
- Ye, C.J.; Sharpe, Z.; Heng, H.H. Origins and Consequences of Chromosomal Instability: From Cellular Adaptation to Genome Chaos-Mediated System Survival. Genes 2020, 11, 1162.
- Baker, S.G. The detached pericyte hypothesis: A novel explanation for many puzzling aspects of tumorigenesis. Org. J. Biol. Sci. 2018, 2, 25–41.
- Vainshelbaum, N.M.; Salmina, K.; Gerashchenko, B.I.; Lazovska, M.; Zayakin, P.; Cragg, M.S.; Pjanova, D.; Erenpreisa, J. Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance. Cells 2022, 11, 880.
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542.
- Ivanov, A.; Cragg, M.S.; Erenpreisa, J.; Emzinsh, D.; Lukman, H.; Illidge, T.M. Endopolyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J. Cell. Sci. 2003, 116, 4095–4106.
- Baker, S.G. Paradoxes in carcinogenesis should spur new avenues of research: An historical perspective. Disrupt. Sci. Technol. 2012, 1, 100–107.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W., Jr. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Baker, S.G. The case for a cancer paradox initiative. Carcinogenesis 2021, 42, 1023–1025.
- Arguello, F. Atavistic Metamorphosis: A New and Logical Explanation for the Origin and Biological Nature of Cancer: With a Discussion on a Novel Approach to Treat Cancer; CreateSpace: Scotts Valley, CA, USA, 2011; ISBN 13:978-1460968994.
- Davies, P. Exposing cancer’s deep evolutionary roots. Phys. World 2013, 26, 37–40.
- Davies, P.C.W.; Lineweaver, C.H. Cancer tumors as Metazoa 1.0: Tapping genes of ancient ancestors. Phys. Biol. 2011, 8, 15001.
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V.; Vazquez-Martin, A.; Salmina, K.; Cragg, M.S. Stress-induced polyploidy shifts somatic cells towards a pro-tumourogenic unicellular gene transcription network. Cancer Hypotheses 2018, 1, 1–20.
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92.
- Erenpreisa, J.; Cragg, M.S. Life-cycle features of tumour cells. In Evolutionary Biology from Concept to Application; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 61–71.
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol. Int. 2005, 29, 1012–1018.
- Erenpreisa, J.; Wheatley, D. Endopolyploidy in development and cancer; “survival of the fattest?”. Cell Biol. Int. 2005, 29, 981–982.
- Lineweaver, C.H.; Davies, P.C.W.; Vincent, M.D. Targeting cancer’s weaknesses (not its strengths): Therapeutic strategies suggested by the atavistic model. BioEssays 2014, 36, 827–835.
- Niculescu, V.F. Developmental and non developmental polyploidy in xenic and axenic cultured stem cell lines of Entamoeba invadens and E. histolytica. Insights Stem. Cells 2016, 2, 1–9.
- Vincent, M.D. Cancer: A de-repression of a default survival program common to all cells? BioEssays 2012, 34, 72–82.
- Vincent, M.D. Cancer: Beyond speciation. Adv. Cancer Res. 2011, 112, 283–350.
- Lineweaver, C.H.; Bussey, K.J.; Blackburn, A.C.; Davies, P.C.W. Cancer progression as a sequence of atavistic reversions. BioEssays 2021, 43, 2000305.
- Yett, J.R. Eukaryotes. In Salem Press Encyclopedia of Science; Grey House Publishing: Amenia, NY, USA, 2015.
- Khozouz, R. Atavistic cancermodel: A new theory of cancer? BioEssays 2021, 43, 2100206.
- Cui, Q.; Wen, S.; Huang, P. Targeting cancer cell mitochondria as a therapeutic approach: Recent updates. Future Med. Chem. 2017, 9, 929–949.
- Kasperski, A.; Kasperska, R. Study on attractors during organism evolution. Sci. Rep. 2021, 11, 9637.
- Kasperski, A.; Kasperska, R. Bioenergetics of life, disease and death phenomena. Theory Biosci. 2018, 137, 155–168.
- Boese, A.C.; Kang, S. Mitochondrial metabolism-mediated redox regulation in cancer progression. Redox Biol. 2021, 42, 101870.
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim. Biophys. Acta 2017, 1867, 151–161.
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, UK, 2000.
- Huang, S.; Ernberg, I.; Kaufman, S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 2009, 20, 869–876.
- Kaufman, S. Diferentiation of malignant to benign cells. J. Theor. Biol. 1971, 31, 429–451.
- Vinogradov, A.E.; Anatskaya, O.V. Evolutionary framework of the human interactome: Unicellular and multicellular giant clusters. Biosystems 2019, 181, 82–87.
- Giuliani, A. Models portability: Some considerations about transdisciplinary approaches, Studies of Nonlinear Phenomena in Life Science. In The Complex Matters of the Mind; World Scientific: Singapore, 1998; pp. 193–210.
- Lewin, R. Complexity: Life at the Edge of Chaos; Maxwell Macmillan International: New York, NY, USA, 1993; ISBN 1857990285.
- Krigerts, J.; Salmina, K.; Freivalds, T.; Zayakin, P.; Rumnieks, F.; Inashkina, I.; Giuliani, A.; Hausmann, M.; Erenpreisa, J. Differentiating cancer cells reveal early large-scale genome regulation by pericentric domains. Biophys. J. 2021, 120, 711–724.
- Zimatore, G.; Tsuchiya, M.; Hashimoto, M.; Kasperski, A.; Giuliani, A. Self-organization of whole-gene expression through coordinated chromatin structural transition. Biophys. Rev. 2021, 2, 031303.
- Zimatore, G.; Tsuchiya, M.; Hashimoto, M.; Kasperski, A.; Giuliani, A. Self-organization of whole gene expression through coordinated chromatin structural transition: Validation of self-organized critical control of genome expression. bioRxiv 2019. bioRxiv: 852681.
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496.
- Liu, G.; Stevens, J.; Horne, S.; Abdallah, B.; Ye, K.; Bremer, S.; Ye, C.; Chen, D.J.; Heng, H. Genome chaos: Survival strategy during crisis. Cell Cycle 2014, 13, 528–537.
- Heng, J.; Heng, H.H. Two-phased evolution: Genome chaos-mediated information creation and maintenance. Prog. Biophys. Mol. Biol. 2021, 165, 29–42.
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2020; in press.
- Heng, H.H. Genome Chaos: Rethinking Genetics, Evolution, and Molecular Medicine; Academic Press Elsevier: Cambridge, MA, USA, 2019; ISBN 978-012-8136-35-5.
- Ye, C.J.; Sharpe, Z.; Alemara, S.; Mackenzie, S.; Liu, G.; Abdallah, B.; Horne, S.; Regan, S.; Heng, H.H. Micronuclei and Genome Chaos: Changing the System Inheritance. Genes 2019, 10, 366.
- Pavlopoulou, A.; Spandidos, D.A.; Michalopoulos, I. Human cancer databases (review). Oncol. Rep. 2015, 33, 3–18.
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome Doubling Shapes the Evolution and Prognosis of Advanced Cancers. Nat. Genet. 2018, 50, 1189–1195.
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with Purpose. Genes Dev. 2009, 23, 2461–2477.
- Pienta, K.J.; Hammarlund, E.U.; Axelrod, R.; Brown, J.S.; Amend, S.R. Poly-Aneuploid Cancer Cells Promote Evolvability, Generating Lethal. Cancer Evol. Appl. 2020, 13, 1626–1634.
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83.
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The Evolutionary Significance of Polyploidy. Nat. Rev. Genet. 2017, 18, 411–424.
- Heng, J.; Heng, H.H. Karyotype coding: The creation and maintenance of system information for complexity and biodiversity. Biosystems 2021, 208, 104476.
- Rancati, G.; Pavelka, N.; Fleharty, B.; Noll, A.; Trimble, R.; Walton, K.; Perera, A.; Staehling-Hampton, K.; Seidel, C.W.; Li, R. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved. Cell 2008, 135, 879–893.
- Duesberg, P.; Rasnick, D. Aneuploidy, the somatic mutation that makes cancer a species of its own. Cell Motil. Cytoskelet. 2000, 47, 81–107.
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Warburg, O.H. The Metabolism of Tumours: Investigations from the Kaiser Wilhelm Institute for Biology; Arnold Constable: Berlin, Germany; Dahlem, Germany; London, UK, 1930.
- Larson, S.M. Positron emission tomography-based molecular imaging in human cancer: Exploring the link between hypoxia and accelerated glucose metabolism. Clin. Cancer Res. 2004, 10, 2203–2204.
- Szigeti, G.P.; Szasz, O.; Hegyi, G. Connections between Warburg’s and Szentgyorgyi’s Approach about the Causes of Cancer. J. Neoplasm 2017, 1, 1–13.
- Blagosklonny, M.V. Molecular theory of cancer. Cancer Biol. Ther. 2005, 4, 621–627.
- Shih, I.M. Gestational trophoblastic neoplasia—Pathogenesis and potential therapeutic targets. Lancet Oncol. 2007, 8, 642–650.
- Shomar, A.; Barak, O.; Brenner, N. Cancer progression as a learning process. Iscience 2022, 25, 103924.
- Greaves, M.; Maley, C. Clonal evolution in cancer. Nature 2012, 481, 306–313.
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312.
- Malaise, E.P.; Chavaudra, N.; Tubiana, M. The relationship between growth rate, labelling index and histological type of human solid tumours. Eur. J. Cancer 1973, 9, 305–312.


