As a phenomenon, cancer is a disease related to multicellular evolution, i.e., cancer in general is understood to be a failure of the multicellular systems and is considered a reversal to unicellularity. Cancer cells are like unicellular organisms that benefit from ancestral-like traits. As a disease, cancer can be interpreted as (a) a destruction of cooperative behaviors underlying multicellular evolution, (b) a disruption of molecular networks established during the emergence of multicellularity or (c) an atavistic state resulting from reactivation of primitive programs typical of the earliest unicellular species. From this point of view and in accordance with the layered model of evolution of cellular functionalities, cancer transformation can occur as a result of huge disturbances or the destruction of functionalities that are located in the multicellular layer.
- cancer
- cancer transformation and development
- universal model of cancer
1. Introduction
2. Cancer Transformation as a Loss of Control over Atavistic Functionalities
Functionalities of the multicellular layer are located in the most external layer of the layered model of evolution of cellular functionalities. In accordance with unified cell bioenergetics, bioenergetic cell problems, especially overenergization of mitochondria, can lead to cancer transformation [28][29]. Cell overenergization (and the related increase in ROS) is followed by adaptation of multiple metabolic strategies to solve this bioenergetic problem [30]. As a result, in light of the layered model of evolution of cellular functionalities, the cell’s response to a huge bioenergetic problem related to overenergization is propagation of disturbances in genome expression from the most internal (i.e., from the layer of bioenergetic functionalities) toward the more external layer (i.e., toward the layer of multicellular functionalities). Functionalities that are localized in the layer of multicellular functionalities, as the most complex and evolutionarily youngest (see Introduction), are the most sensitive to disturbances. The disturbance (or destruction, for example by, high ROS levels) of multicellular layer functionalities can result in a loss of control over functionalities of the unicellular layer, leading to cancer transformation, i.e., uncontrolled activity of atavistic functionalities.3. Vertical and Horizontal Cancer Development
Results of studies on tumor development published to date (deep sequencing, multi-region sequencing and single-cell sequencing) indicate that a single normal cell is a common origin of cancer [31]. In order to increase the probability of survival, cancer transformation initiates the creation of multiple clones, along with activation of the ploidy cycle (see Introduction) [6][32]. Transformation also initiates cancer development, which, according to the universal model of cancer transformation and development (Figure 1), occurs as the development of a population of individual cloning cells. Due to the very high complexity of the task, establishing a universal model of cancer transformation and development can be treated as a process of successive extensions and improvements. The first approaches to establishing such a universal model were presented in previously published articles [28][29]. The universal model of cancer transformation and development (Figure 1) is presented from the perspective of the proposed layered model of evolution of cellular functionalities (especially from the perspective of losing control over functionalities of the unicellular layer), which can be considered a significant extension and improvement compared to the previously presented approaches. Figure 1, with a very synthetic explanation (compared to that previously presented in Figure 7 in [28] and Figure 3 in [29]) allows for a more complete understanding of the presented considerations. According to the universal model of cancer transformation and development, vertical and optional horizontal development can be distinguished [28]. Both vertical and horizontal cancer development occur through step-by-step changes of attractors, i.e., caner development shows attractor-like behavior. The idea of “cancer attractors” was first proposed by Stuart Kaufman and has been supported experimentally [33][34]. The use of the attractor idea has made it possible to explain why the cells of multicellular organisms are prone to oncogenesis [35]. In general, the application of the attractor concept allows the physical concept to be used in biological reality, where the term “attractor” denotes the configuration towards which the system evolves over time to achieve stability [36][37]. Attaining an attractor means that a given system configuration is stable enough to return to its original state after the disappearance of any small disturbances [37]. Additionally, in Figure 1, exemplary attractors of normal cell fates are marked on the vertical green axis. It should be noted that all normal cell fates are generated by cells trapped in one genome attractor. This is because genome DNA sequences in each cell nucleus are identical in human cells (and tissues of each individual) [38].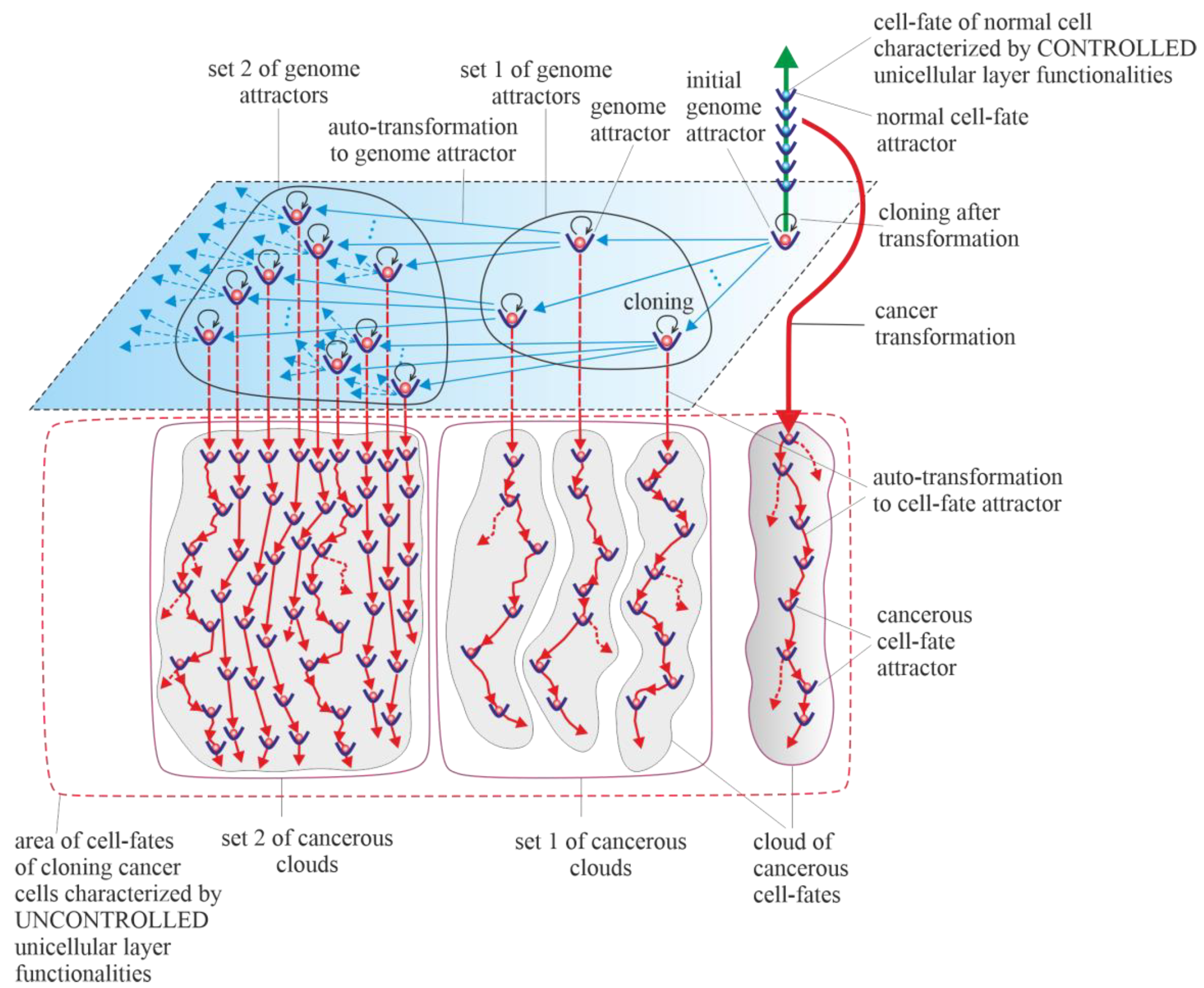
4. Vertical Cancer Development
Vertical cancer development occurs when cells change cell-fate attractors without a change in genome attractor, and for this reason, this type of cancer development can be considered a kind of microevolution. A change in cell-fate attractor can occur as a result of the occurrence of considerable instability of genome expression (i.e., considerable instability of current cell fate) [39][40]. Vertical cancer development can occur without mutations (for example, only as a result of cell bioenergetic problems) or with mutations as an associated phenomenon (but under the condition that these mutations do not cause leaving of the genome attractor). Autotransformation to cell-fate attractor (i.e., transformation that constitutes an ordered cell response to cell-fate instability) causes ordered changes of genome expression introduced in order to attain cell-fate stability [28]. That means that autotransformation to the cell-fate attractor causes stabilization of cell-fate in the new cell-fate attractor. As has been presented in published articles, activation and stabilization of new cell-fate occurs by positional chromatin remodeling [39][40].5. Horizontal Cancer Development
Horizontal cancer development occurs when cells change genome attractors, and for this reason, this type of cancer development can be considered a kind of macroevolution. It should be added horizontal cancer development is optional; it may occur or it may not [28]. Change in genome attractor can occur as a result of a loss of genome stability. Destabilization of the genome can follow destruction of the DNA fragments that code mechanisms responsible for monitoring genomic integrity by random mutations caused by a high level of ROS [41]. The destruction of these mechanisms and consequent loss of genomic integrity can lead to genome instability (GIN) and, as a result, to genome chaos. Genome chaos is a process of complex, rapid genome reorganization that results in the formation of unstable genomes [42]. These unstable genomes have the potential to establish stable genomes [42]. Taking into account that during cancer development, adaptation of clones to stress occurs by the production of new genomes that are essential for phase transition, the occurrence of genome chaos is an important factor in cancer development [1][43][44][45][46]. The aim of autotransformation to the genome attractor (which constitutes an ordered cell response to the formation of an unstable genome as a result of occurrence of genome chaos) is stabilization of unstable genomes [28]. Autotransformation to the genome attractor causes unstable genome changes (including aneuploidy, rearrangements and other ordered genome changes) introduced in order to attain genomic stability. That means that autotransformation to the attractor causes stabilization of the genome in a new genome attractor. Attaining a new genome attractor has to be followed by a change of cell fate in order to keep the cell alive by adjusting genome expression to the changed genome. That means that autotransformation to the attractor has to be followed by autotransformation to the cell-fate attractor [28]. From this point of view, cancer development occurs as a kind of process of self-creation, i.e., “under high-stress conditions likely to eliminate a system, the system’s cellular machinery will automatically switch into a mode that destroys the current genome and simultaneously forms new genomes using their own genomic materials” [1]. The aim of cancer development as a process (including, among other factors, successive losses of genomic integrity that lead to subsequent genome instability (GIN), genome chaos, formation of unstable genomes followed by autotransformation to the genome attractor and autotransformation to the cell-fate attractor) is to form new genomes and keep cells alive. Cancer is characterized by abundant genetic abnormalities in the form of mutations, single-nucleotide polymorphisms, copy-number alterations, genomic rearrangements and gene fusions [47]. Moreover, aneuploidy appears early during cancer development and correlates with cancer aggression and resistance to anticancer treatments, favoring cancer progression and poor prognosis [6][48][49][50][51][52]. Resistance to anticancer treatments may be related to the possibility of recovering an individual gene’s function by aneuploidy [53][54]. Cancers are known to be clonal for aneuploidy above a certain threshold [55]. Aneuploidy is a phenomenon associated with horizontal cancer development. In accordance with the universal model of cancer transformation and development, horizontal cancer development causes a perpetual increase in aneuploidy, along with permanent cloning, when aneuploidy passes the threshold. A change in the karyotype (i.e., a change in the whole sets of functionalities) related to the addition or removal of one small chromosome can alter overall gene expression [53][54]. That means that aneuploidy has a large impact on cancer development, especially on genome and cell-fate heterogeneity. Vertical and horizontal cancer development allows the cancer cells to test the genomic landscape. Horizontal development as macroevolution is associated by large changes in the genome and allows for simultaneous testing of the genomic landscape in many evolutionarily distant places (i.e., horizontal development allows for global testing of the genomic landscape). Vertical development as microevolution is associated with development without changes or small changes in the genome that do not cause changes in the genome attractor. Vertical cancer development confirms whether it is possible to keep clones alive (by changing cell fates appropriately) in distant places (established by horizontal development), i.e., in genome attractors. Additionally, vertical development allows clones to adapt to the environment by cell-fate changes. An exact adaptation to environment can occur by local testing of the genomic landscape, i.e., by cloning and small changes in the genome (caused by high ROS levels) and, related to these changes, changes in cell fates. In this way, adjusting of cell fates to extracellular and intracellular conditions can occur. Horizontal cancer development can be compared to sowing different seeds (seeds represent clones) in a large field. Vertical cancer development can be compared to checking whether it is possible to maintain life in given places in the field and to adjusting life to these places. The information presented so far can be summarized as follows:- (a) Cancer transformation is caused by a loss of control over atavistic functionalities;
- (b) Vertical cancer development is caused by recurring (repeated) losses of current cell-fate stability; at
- (c) Horizontal cancer development is optional and is caused by recurring (repeated) losses of genomic integrity.
6. Cancerous Clouds of Atavistic Cell-Fates
The Warburg effect, a phenomenon associated with cancer transformation and development, causes intensive stimulation of fermentation under good aerobic conditions [56][57][58]. Intensive fermentation allows cells to obtain needed energy during glycolysis. The rate of energy attainment after cancer transformation from intensive glycolysis can be higher than the rate of energy attainment from oxidative phosphorylation (OXPHOS). OXPHOS is a very effective way to obtain energy, but taking into account that a possible fermentation rate can be 100 times quicker than the oxidative process of ATP generation by mitochondria, the rate of energy attainment from glycolysis (i.e., in the glycolysis-fermentation pathway) can be about six times higher in comparison to OXPHOS [59][60]. After transformation, the large amount of obtained energy in the glycolysis-fermentation pathway prevents discharge of mitochondria from high-energy molecules [29]. As a result, this phenomenon causes cancer mitochondria to remain overenergized. Overenergization, as an internal cell problem, is a factor that drives cancer vertical development (i.e., cancer microevolution) and cancer horizontal development (i.e., cancer macroevolution). A large amount of obtained energy after transformation also allows for intensive cell cloning, which creates a network of cell fates (herein termed “cloud of cell-fates”). A cloud of cell fates is generated by cells initially trapped in one (initial) genome attractor. Considering the case of horizontal cancer development, the network of genome attractors undergoes gradual and dynamic expansion. As a result, sets of cancerous clouds (that contain cancerous clouds of atavistic cell fates) are generated by cloning cells trapped in a dynamically emerging and altered network of genome attractors.7. Purely Vertical Cancer Development
Horizontal cancer development is optional (i.e., it may occur or not). From this point of view, a special case of vertical cancer development is vertical development, with no one change of genome attractor (i.e., purely vertical cancer development). This means that during cancer development, cancer clones are trapped and kept in the initial genome attractor. The initial genome attractor is a genome attractor of a normal cell in which the cell has been transformed into a cancerous cell (see Section 3.2). After transformation, cancer development occurs as subsequent changes in cell-fate attractors due to destabilizations of current cell fates. Destabilization of cell fates can occur as a result of bioenergetic problems (i.e., without mutations) and as a result of mutations that do not cause a change in genome attractor.8. Cancers without Mutation
Teratomas and choriocarcinomas, as exemplary cancers without mutations, are formed by misplaced embryonic and placental cells [61]. These cells are characterized by altered expression of hundreds of oncogenes and tumor suppressors (silenced or induced by epigenetic mechanisms) compared to adult tissues [61]. In choriocarcinoma, HLA-G is demonstrated to change the tumor microenvironment through the inactivation of the local immune system at very high levels and functions [62]. Moreover, choriocarcinoma is characterized by overexpression of p53 and MDM2, along with overexpression of other genes (i.e., NECC1, epidermal growth factor receptor, DOC-2/hDab2, Ras GTPase-activating protein, E-cadherin, HIC-1, p16 and TIMP3) or downregulation via hypermethylation, with no evidence of somatic mutation [62]. In this light, a special case of vertical cancer development is purely vertical cancer development without any mutation. That means that during cancer development, cancer clones are trapped and kept in the initial genome attractor, and additionally, cancer development occurs without mutations (i.e., only as a result of bioenergetic problems that lead to subsequent destabilizations of cell fates and changes in cell-fate attractors).9. Cancer Development as a Learning Process
Cancer progression can be considered a learning process [63]. This learning process is very costly because a large number of cancerous cells dies during cancer development [64][65][66]. Taking into account that the cancer cell-doubling time is around 1–2 days and that tumor-doubling time is around 60–200 days, the conclusion is that the vast majority of cancer cells die before they can divide [64][65][66]. Even advanced malignancies can exhibit such characteristics of growth (i.e., Gompertzian growth) [65]. In light of the universal model of cancer transformation and development, horizontal cancer development is driven by successive destructions of mechanisms responsible for monitoring genomic integrity. Consequently, a loss of genomic integrity can lead to genome instability (GIN) and, as a result, genome chaos and formation of unstable genomes (as an outcome of genomic chaos). Taking into account cancer Gompertzian growth, in light of the universal model of cancer transformation and development, only a small part of the unstable genomes can undergo transformation (by autotransformation to the genome attractor) to stable genomes and attain stability in genome attractors. It should also be taken into account that not all stable genomes have the potential to keep the cell alive. This means that only part of the clones with stable genomes can undergo successful vertical transformation and attain cell-fate stability in cell-fate attractors.References
- Heng, J.; Heng, H.H. Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”. Genes 2022, 13, 101.
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of cancer: Survival at the brink. Semin. Cancer Biol. 2020; in press.
- Heng, H.H. Debating Cancer: The Paradox in Cancer Research; World Scientific Publishing Co.: Singapore, 2015; ISBN 978-981-4520-84-3.
- Ye, C.J.; Sharpe, Z.; Heng, H.H. Origins and Consequences of Chromosomal Instability: From Cellular Adaptation to Genome Chaos-Mediated System Survival. Genes 2020, 11, 1162.
- Baker, S.G. The detached pericyte hypothesis: A novel explanation for many puzzling aspects of tumorigenesis. Org. J. Biol. Sci. 2018, 2, 25–41.
- Vainshelbaum, N.M.; Salmina, K.; Gerashchenko, B.I.; Lazovska, M.; Zayakin, P.; Cragg, M.S.; Pjanova, D.; Erenpreisa, J. Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance. Cells 2022, 11, 880.
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542.
- Ivanov, A.; Cragg, M.S.; Erenpreisa, J.; Emzinsh, D.; Lukman, H.; Illidge, T.M. Endopolyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J. Cell. Sci. 2003, 116, 4095–4106.
- Baker, S.G. Paradoxes in carcinogenesis should spur new avenues of research: An historical perspective. Disrupt. Sci. Technol. 2012, 1, 100–107.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W., Jr. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Baker, S.G. The case for a cancer paradox initiative. Carcinogenesis 2021, 42, 1023–1025.
- Arguello, F. Atavistic Metamorphosis: A New and Logical Explanation for the Origin and Biological Nature of Cancer: With a Discussion on a Novel Approach to Treat Cancer; CreateSpace: Scotts Valley, CA, USA, 2011; ISBN 13:978-1460968994.
- Davies, P. Exposing cancer’s deep evolutionary roots. Phys. World 2013, 26, 37–40.
- Davies, P.C.W.; Lineweaver, C.H. Cancer tumors as Metazoa 1.0: Tapping genes of ancient ancestors. Phys. Biol. 2011, 8, 15001.
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V.; Vazquez-Martin, A.; Salmina, K.; Cragg, M.S. Stress-induced polyploidy shifts somatic cells towards a pro-tumourogenic unicellular gene transcription network. Cancer Hypotheses 2018, 1, 1–20.
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92.
- Erenpreisa, J.; Cragg, M.S. Life-cycle features of tumour cells. In Evolutionary Biology from Concept to Application; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 61–71.
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol. Int. 2005, 29, 1012–1018.
- Erenpreisa, J.; Wheatley, D. Endopolyploidy in development and cancer; “survival of the fattest?”. Cell Biol. Int. 2005, 29, 981–982.
- Lineweaver, C.H.; Davies, P.C.W.; Vincent, M.D. Targeting cancer’s weaknesses (not its strengths): Therapeutic strategies suggested by the atavistic model. BioEssays 2014, 36, 827–835.
- Niculescu, V.F. Developmental and non developmental polyploidy in xenic and axenic cultured stem cell lines of Entamoeba invadens and E. histolytica. Insights Stem. Cells 2016, 2, 1–9.
- Vincent, M.D. Cancer: A de-repression of a default survival program common to all cells? BioEssays 2012, 34, 72–82.
- Vincent, M.D. Cancer: Beyond speciation. Adv. Cancer Res. 2011, 112, 283–350.
- Lineweaver, C.H.; Bussey, K.J.; Blackburn, A.C.; Davies, P.C.W. Cancer progression as a sequence of atavistic reversions. BioEssays 2021, 43, 2000305.
- Yett, J.R. Eukaryotes. In Salem Press Encyclopedia of Science; Grey House Publishing: Amenia, NY, USA, 2015.
- Khozouz, R. Atavistic cancermodel: A new theory of cancer? BioEssays 2021, 43, 2100206.
- Cui, Q.; Wen, S.; Huang, P. Targeting cancer cell mitochondria as a therapeutic approach: Recent updates. Future Med. Chem. 2017, 9, 929–949.
- Kasperski, A.; Kasperska, R. Study on attractors during organism evolution. Sci. Rep. 2021, 11, 9637.
- Kasperski, A.; Kasperska, R. Bioenergetics of life, disease and death phenomena. Theory Biosci. 2018, 137, 155–168.
- Boese, A.C.; Kang, S. Mitochondrial metabolism-mediated redox regulation in cancer progression. Redox Biol. 2021, 42, 101870.
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim. Biophys. Acta 2017, 1867, 151–161.
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, UK, 2000.
- Huang, S.; Ernberg, I.; Kaufman, S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 2009, 20, 869–876.
- Kaufman, S. Diferentiation of malignant to benign cells. J. Theor. Biol. 1971, 31, 429–451.
- Vinogradov, A.E.; Anatskaya, O.V. Evolutionary framework of the human interactome: Unicellular and multicellular giant clusters. Biosystems 2019, 181, 82–87.
- Giuliani, A. Models portability: Some considerations about transdisciplinary approaches, Studies of Nonlinear Phenomena in Life Science. In The Complex Matters of the Mind; World Scientific: Singapore, 1998; pp. 193–210.
- Lewin, R. Complexity: Life at the Edge of Chaos; Maxwell Macmillan International: New York, NY, USA, 1993; ISBN 1857990285.
- Krigerts, J.; Salmina, K.; Freivalds, T.; Zayakin, P.; Rumnieks, F.; Inashkina, I.; Giuliani, A.; Hausmann, M.; Erenpreisa, J. Differentiating cancer cells reveal early large-scale genome regulation by pericentric domains. Biophys. J. 2021, 120, 711–724.
- Zimatore, G.; Tsuchiya, M.; Hashimoto, M.; Kasperski, A.; Giuliani, A. Self-organization of whole-gene expression through coordinated chromatin structural transition. Biophys. Rev. 2021, 2, 031303.
- Zimatore, G.; Tsuchiya, M.; Hashimoto, M.; Kasperski, A.; Giuliani, A. Self-organization of whole gene expression through coordinated chromatin structural transition: Validation of self-organized critical control of genome expression. bioRxiv 2019. bioRxiv: 852681.
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496.
- Liu, G.; Stevens, J.; Horne, S.; Abdallah, B.; Ye, K.; Bremer, S.; Ye, C.; Chen, D.J.; Heng, H. Genome chaos: Survival strategy during crisis. Cell Cycle 2014, 13, 528–537.
- Heng, J.; Heng, H.H. Two-phased evolution: Genome chaos-mediated information creation and maintenance. Prog. Biophys. Mol. Biol. 2021, 165, 29–42.
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2020; in press.
- Heng, H.H. Genome Chaos: Rethinking Genetics, Evolution, and Molecular Medicine; Academic Press Elsevier: Cambridge, MA, USA, 2019; ISBN 978-012-8136-35-5.
- Ye, C.J.; Sharpe, Z.; Alemara, S.; Mackenzie, S.; Liu, G.; Abdallah, B.; Horne, S.; Regan, S.; Heng, H.H. Micronuclei and Genome Chaos: Changing the System Inheritance. Genes 2019, 10, 366.
- Pavlopoulou, A.; Spandidos, D.A.; Michalopoulos, I. Human cancer databases (review). Oncol. Rep. 2015, 33, 3–18.
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome Doubling Shapes the Evolution and Prognosis of Advanced Cancers. Nat. Genet. 2018, 50, 1189–1195.
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with Purpose. Genes Dev. 2009, 23, 2461–2477.
- Pienta, K.J.; Hammarlund, E.U.; Axelrod, R.; Brown, J.S.; Amend, S.R. Poly-Aneuploid Cancer Cells Promote Evolvability, Generating Lethal. Cancer Evol. Appl. 2020, 13, 1626–1634.
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83.
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The Evolutionary Significance of Polyploidy. Nat. Rev. Genet. 2017, 18, 411–424.
- Heng, J.; Heng, H.H. Karyotype coding: The creation and maintenance of system information for complexity and biodiversity. Biosystems 2021, 208, 104476.
- Rancati, G.; Pavelka, N.; Fleharty, B.; Noll, A.; Trimble, R.; Walton, K.; Perera, A.; Staehling-Hampton, K.; Seidel, C.W.; Li, R. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved. Cell 2008, 135, 879–893.
- Duesberg, P.; Rasnick, D. Aneuploidy, the somatic mutation that makes cancer a species of its own. Cell Motil. Cytoskelet. 2000, 47, 81–107.
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Warburg, O.H. The Metabolism of Tumours: Investigations from the Kaiser Wilhelm Institute for Biology; Arnold Constable: Berlin, Germany; Dahlem, Germany; London, UK, 1930.
- Larson, S.M. Positron emission tomography-based molecular imaging in human cancer: Exploring the link between hypoxia and accelerated glucose metabolism. Clin. Cancer Res. 2004, 10, 2203–2204.
- Szigeti, G.P.; Szasz, O.; Hegyi, G. Connections between Warburg’s and Szentgyorgyi’s Approach about the Causes of Cancer. J. Neoplasm 2017, 1, 1–13.
- Blagosklonny, M.V. Molecular theory of cancer. Cancer Biol. Ther. 2005, 4, 621–627.
- Shih, I.M. Gestational trophoblastic neoplasia—Pathogenesis and potential therapeutic targets. Lancet Oncol. 2007, 8, 642–650.
- Shomar, A.; Barak, O.; Brenner, N. Cancer progression as a learning process. Iscience 2022, 25, 103924.
- Greaves, M.; Maley, C. Clonal evolution in cancer. Nature 2012, 481, 306–313.
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312.
- Malaise, E.P.; Chavaudra, N.; Tubiana, M. The relationship between growth rate, labelling index and histological type of human solid tumours. Eur. J. Cancer 1973, 9, 305–312.