Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Edward Prochownik | -- | 4500 | 2022-04-12 14:47:58 | | | |
| 2 | Catherine Yang | + 2 word(s) | 4502 | 2022-04-13 03:02:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Prochownik, E.; Wang, H. Extended Myc Network. Encyclopedia. Available online: https://encyclopedia.pub/entry/21657 (accessed on 26 July 2026).
Prochownik E, Wang H. Extended Myc Network. Encyclopedia. Available at: https://encyclopedia.pub/entry/21657. Accessed July 26, 2026.
Prochownik, Edward, Huabo Wang. "Extended Myc Network" Encyclopedia, https://encyclopedia.pub/entry/21657 (accessed July 26, 2026).
Prochownik, E., & Wang, H. (2022, April 12). Extended Myc Network. In Encyclopedia. https://encyclopedia.pub/entry/21657
Prochownik, Edward and Huabo Wang. "Extended Myc Network." Encyclopedia. Web. 12 April, 2022.
Copy Citation
Among the first discovered and most prominent cellular oncogenes is MYC, which encodes a bHLH-ZIP transcription factor (Myc) that both activates and suppresses numerous genes involved in proliferation, energy production, metabolism and translation. Myc belongs to a small group of bHLH-ZIP transcriptional regulators (the Myc Network) that includes its obligate heterodimerization partner Max and six “Mxd proteins” (Mxd1–4, Mnt and Mga), each of which heterodimerizes with Max and largely opposes Myc’s functions.
L-Myc
Max
Mxd Cancer Metabolism Tumor suppressor ChREBP MondoA Mlx
1. Introduction
The Myc and Mlx Networks. The year 2021 marked the 30th anniversary of the discovery of Max, the obligate heterodimerization partner of the c-Myc (Myc) bHLH-ZIP transcription factor, which had itself been identified nearly a decade earlier as the evolutionarily conserved cellular homolog of the avian v-myc retroviral oncogene [1][2][3][4][5]. Shortly thereafter, it became clear that Max was necessary for Myc-mediated target gene activation and cellular transformation but that, at higher Max:Myc ratios, it also repressed these functions in a dose-dependent manner [6][7][8][9][10][11]. This initially suggested a simple model, whereby Myc–Max heterodimers, which contain a transactivation domain (TAD) contributed by Myc, and Max homodimers, which lack a TAD, alternatively activate or suppress transcription in accord with their relative abundance [12][13]. This balance was believed to be maintained by the competitive binding of these dimers to so-called “E-box” elements that are typically located in proximity to the transcriptional start sites of target genes (Figure 1). The model immediately implied that Max could serve dual functions—on the one hand, it could activate target genes and drive transformation by virtue of its obligate association with Myc; as a homodimer, on the other hand, it could outcompete Myc–Max binding, repress target gene expression and serve as a tumor suppressor (TS).
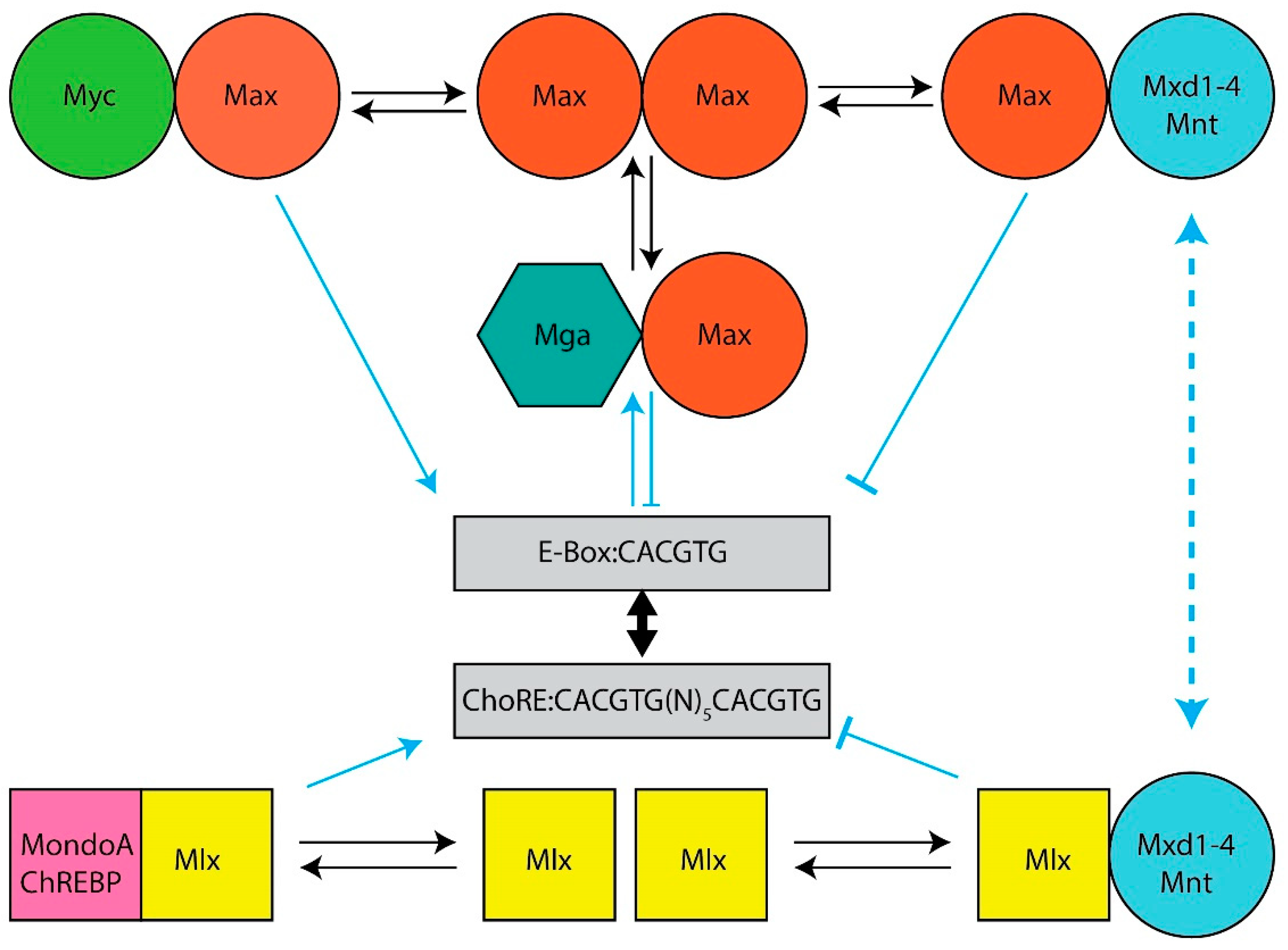
Figure 1. The Extended Myc Network. Top: The Myc Network. Myc–Max heterodimers bind to cognate E-boxes in target genes. They then recruit chromatin-modifying enzymes such as histone acetyl transferases and transcription co-factors, which mediate the histone acetylation and demethylation that permit subsequent increases in RNA polymerase II (Pol II) binding, the relief of Pol II pausing and read-through transcription [14][15][16][17][18]. DNA binding by Max homodimers is prevented by inhibitory N-terminal phosphorylation [19][20]. Max also lacks a TAD that is needed for chromatin remodeling and transcriptional activation [13][16][21]. Transcriptional repression of Myc-activated genes is mediated by six “Mxd proteins” (Mxd1–4, Mnt and Mga) whose levels of expression are variably tissue, development, age and cell cycle specific [22][23][24][25][26][27]. These compete with Myc–Max heterodimers for E-boxes, recruit mSin3, histone deacetylases, methyltransferases and complexes that mediate more direct transcriptional repression [28][29][30]. Bottom: The Mlx Network. The Myc-like factors MondoA and ChREBP heterodimerize with Mlx (which, unlike Max, can homodimerize, albeit weakly), bind certain metabolites such as glucose, glucose-6-phosphate, lactate and adenosine, and translocate from the cytoplasm to the nucleus where they bind to target genes containing both ChoREs and E-boxes although the size of this repertoire is smaller than that of Myc targets [31][32][33][34][35][36][37][38][39][40][41][42]. The negative regulatory arm of the Mlx Network employs some of the same repressive strategies utilized by the Myc Network with some distinct exceptions.
Max’s discovery soon paved the way for the rapid identification of six additional bHLH-ZIP Max partners that currently comprise the so-called “Myc Network”, of which Max remains the central member (Figure 1). These proteins, Mxd1–4, Mnt and Mga (hereafter referred to collectively as “Mxd proteins”, despite their structural and functional differences) (Figure 1), necessitated a revision of the above model as did the finding that, in mammalian cells, Max’s high level of phosphorylation, maintained by casein kinase II, prevents its binding to DNA as a homodimer but not as a heterodimer [19][20][43]. Transcriptional repression by Max was thus deemed to be mediated not simply by passive exclusion of Myc–Max heterodimers from binding E-boxes but as an active process that required Max’s heterodimerization with Mxd proteins, the association with obligate transcriptional co-repressors such as mSin3 proteins and the ensuing transcriptionally suppressive modification of chromatin via altered patterns of histone acetylation and methylation [14][28][44][45][46][47]. Collectively, these six Mxd proteins, which show distinct tissue-, developmental- and age-dependent patterns of expression, antagonize Myc’s broad impact on transcription and its highly pleiotropic effects on both normal and neoplastic growth [6][7][8][22][24][46][48][49][50][51][52][53][54][55].
By the turn of the 21st century, the existence of a second network, parallel to that of the Myc Network, and possessing significant structural and functional similarities, had emerged. This “Mlx Network”, also comprised of bHLH-ZIP factors, contained the Myc-like proteins ChREBP (“carbohydrate response element binding protein”) and MondoA and their own dedicated heterodimerization partner, the Max-related Mlx, which together formed the Network’s positively-acting arm [31][56][57][58] (Figure 1). Unlike Myc and Max, which are nuclear proteins, ChREBP, MondoA and Mlx are “conditionally nuclear” in that they translocate to the nucleus only upon binding metabolites such as glucose, glucose-6-phosphate, fructose 2,6-bisphosphate, lactate and adenosine [31][32][33][34][35][36][37][38][40][42][56][59][60][61][62][63][64][65][66][67][68]. The cytosolic location of MondoA and ChREBP is also not random; rather, amphipathic helical domains in the C-termini of these factors allow them to interact with intracytoplasmic lipid droplets and presumably serve as sensors of intracellular lipid content [69]. Lipid droplet depletion, together with the above metabolites, allows MondoA and ChREBP to translocate to the nucleus and activate genes involved in glucose metabolism. The negative arm of the Mlx Network shares three factors from the Myc Network, namely Mxd1, Mxd4 and Mnt, along with mSin3–histone deacetylase complexes [31][57][70]. These three negative regulators link the two Networks both structurally and functionally and provide a means of cross-talk for more refined and coordinated regulation of both common and unique targets.
Mlx Network heterodimers recognize E-box-related binding sites termed “carbohydrate response elements” (ChoREs) (Figure 1). Both E-boxes and ChoREs, but particularly the latter, can be quite variable and cross-binding of one Network’s members to the other Network’s sites is likely more common than previously appreciated [57][71][72][73][74][75][76]. Binding by ChREBP may also facilitate promiscuous, non-E-box-dependent binding by Myc at nearby sites [77]. Mlx Network target genes, while overlapping with those regulated by the Myc Network, are both fewer in number and have been reported to be more functionally restricted [39][41][78]. They tend to encode enzymes involved in carbohydrate and lipid metabolism; mitochondrial and ribosomal proteins and factors that regulate translational initiation, elongation and termination [31][39][41][42][46][70][71][72][73][76][78][79][80][81][82][83][84][85][86][87][88][89]. Together, these findings indicate that, via selective, cytoplasmic-nuclear partitioning, sharing of Mxd1,4 and Mnt and binding to both common and unique target genes, this “Extended Myc Network” cross-talks, while simultaneously communicating with and responding to the metabolic cues and reservoirs needed to support energy-intensive processes such as translation and proliferation [36][42][64][65][69][88][89][90][91][92][93][94][95][96].
The Extended Myc Network and tumor suppression. Given the number of Mxd proteins and their crucial roles, particularly with regard to limiting Myc signaling, proliferation and metabolism (Figure 1), it is reasonable to surmise that, at some level, they, and perhaps Max as well, behave as TSs. The researchers discuss below the evidence to support this, the findings that the inactivation of these factors is often confined to certain tumor types and the implications of Mxd member haplo-insufficiency, which is a common theme in many tumors. It is also important to consider these factors in their proper biological context. In other words, do they behave as “classical” TSs such TP53, RB and BRCA1/2, whose germ line or somatic inactivation predispose to spontaneous neoplasms such as leukemias, lymphomas, retinoblastomas, osteosarcomas, and breast, ovarian and prostate cancer [97][98][99][100][101][102]? Or, does the inactivation of these factors simply accelerate the growth of pre-existing tumors without otherwise contributing directly to their initiation?
All Mxd members, as well as the central players Max and Mlx, have been implicated as TSs based on bioinformatics-based surveys of large populations of human tumors [103][104]. However, to our knowledge, neither comprehensive summaries nor intragroup comparisons of the consequences of copy number variations (CNVs) and mutations described in these reports have been published. By way of introduction to the more detailed discussions presented in the following sections, the researchers surveyed all tumors from The Cancer Genome Atlas (TCGA) to identify those associated with CNVs of Extended Myc Network genes (Figure 2). This survey revealed several interesting findings. First, and in keeping with the known role of MYC as a bona fide oncogene and the presumptive roles of CHREBP and MONDOA in supporting transformation and rapid proliferation, these genes showed the highest frequency of amplification [36][41][45][103][104]. Second, recurrent deletions of all other Extended Myc Network members were quite frequent across many tumor types, in keeping with their presumptive role as TS genes (TSGs). Third, none of the genes was exclusively amplified or deleted across all tumor types or, in many cases, even within the same tumor type [104]. Fourth, among the “oncogenes”, high-level gene amplification, defined as >4.4 copies/cell [105], was seen only with MYC and tended to be associated with subgroups of certain cancers such as breast, ovary, liver and lung as previously reported [106][107][108][109]. Fifth, more often than not, amplifications or deletions of more than one Extended Myc Network gene were associated with particular tumor types and were most obvious in breast, ovarian and squamous cell lung cancers. This suggests that oncogenic activation of the Extended Myc Network can be achieved via multiple pathways that are cooperative rather than mutually exclusive. Sixth, despite its relatively high incidence of genomic instability, thyroid cancer was the only tumor in which CNV of any Extended Myc Network member was never observed (Figure 2). This may partly reflect the tumor’s high incidence of oncogenic BRAF, PIK3CA and PTEN mutations, which likely circumvent Myc Network activation [104][110]. In keeping with the theme that high-level MYC amplification tends to be confined to certain tumor types (Figure 2) [104], deletion of presumptive TSGs tend to cluster as well, particularly in those instances associated with two copy deletion as seen with MLX and MNT in ovarian cancer, MXD2 in prostate cancer and MXD3 in squamous cell lung cancer (Figure 2) [104]. Finally, the inactivation of TSG-like members of the Extended Myc Network by frame-shift and truncating mutations is rare, typically accounting for <1% of inactivated alleles, except in the case of MGA, where this approaches 4% [104]. Thus, as discussed more fully in the following sections, rather than the traditional bi-allelic inactivation/deletion of classical TSGs seen in both hereditary and sporadic cancers, hemizygous inactivation of MXD genes is not only common but a recurrent theme [98][99][100][101][102].

Figure 2. CNVs among individual members of the Extended Myc Network. Gene-level copy number (gistic2_thresholded) [111] data were downloaded from the TCGA Pan-Cancer (PANCAN) database (https://xenabrowser.net/datapages/?cohort=TCGA%20Pan-Cancer %20(PANCAN), accessed on 15 November 2021) Heatmaps were drawn using ComplexHeatmap R package [112]. Tumor type abbreviations: ACC: adrenocortical carcinoma; BLCA: bladder urothelial carcinoma; BRCA: breast invasive carcinoma; CESC: cervical squamous cell carcinoma/endocervical adenocarcinoma; CHOL: cholangiocarcinoma; COAD: colon adenocarcinoma; DLBC: diffuse large B-cell lymphoma; ESCA: esophageal carcinoma; GBM: glioblastoma multiforme; HNSC: head and neck squamous cell carcinoma; KICH: kidney chromophobe carcinoma; KIRC: kidney clear cell carcinoma; KIRP: kidney renal papillary cell carcinoma; LAML: acute myeloid leukemia; LGG: lower-grade glioma; LIHC: hepatocellular carcinoma; LUAD: lung adenocarcinoma; LUSC: lung squamous cell carcinoma; MESO: mesothelioma; OV: ovarian serous cystadenocarcinoma; PAAD: pancreatic adenocarcinoma; PCPG: pheochromocytoma/paraganglioneuroma; PRAD: prostate adenocarcinoma; READ: rectal adenocarcinoma; SARC: sarcoma; SKCM: skin cutaneous melanoma; STAD: stomach adenocarcinoma; TGCT: testicular germ cell tumor; THCA: thyroid carcinoma; THYM: thymoma; UCEC: uterine corpus endometrial carcinoma; UCS: uterine carcinosarcoma; UVM: uveal melanoma.
As was generally true for oncogenic members of the Extended Myc Network, the correlation between MXD TSG expression and survival is variable. Figure 3 summarizes these relationships among all tumors from TCGA and Figure 4 shows several specific examples of survival differences. Interestingly, survival correlated with the expression of as many as 8–9 of the individual Extended Myc Network members and in the cases of low-grade gliomas (LGGs) and clear cell renal cancers (KIRC), with all 11 members, although not necessarily in the same way. This supports the points mentioned above that perturbing the delicate balance among Extended Myc Network members may have oncogenic consequences that are additive or complementary rather than mutually exclusive. The connections among tumor aggressiveness, survival and the expression of individual members of the Extended Myc Network are thus complex, interdependent and presumably tumor type specific.
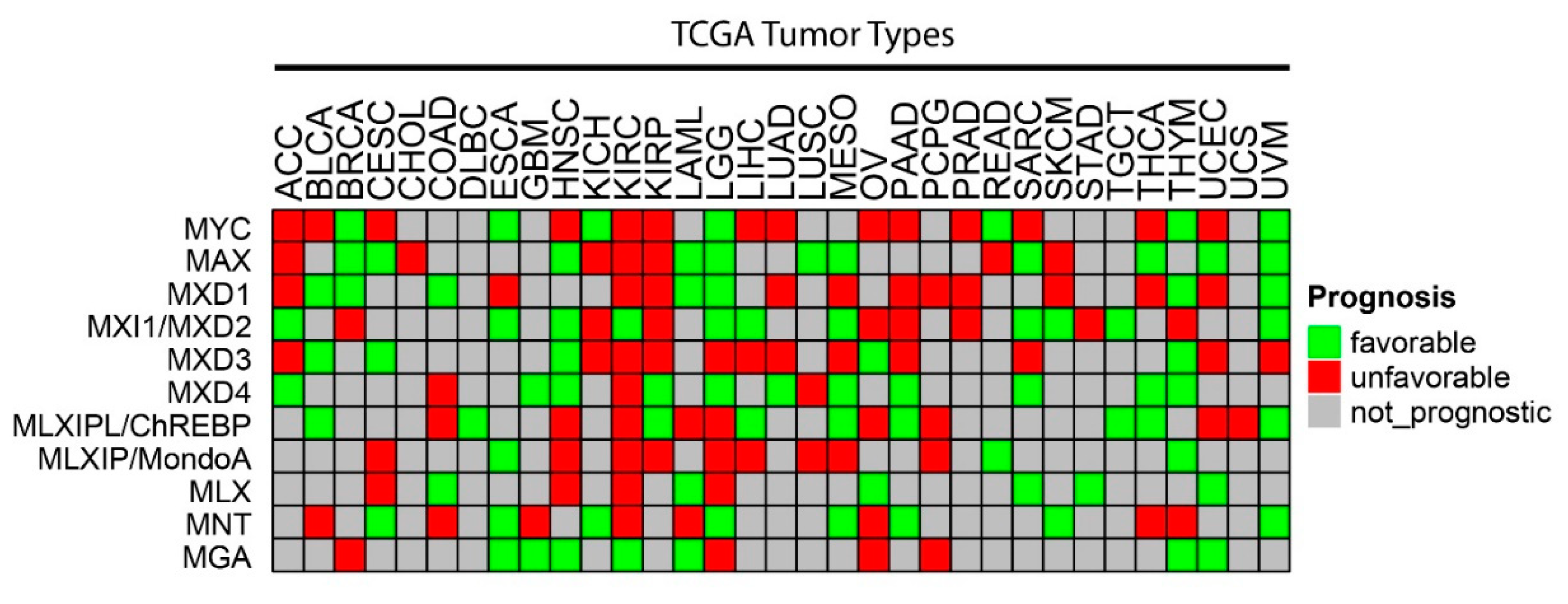
Figure 3. Correlations between survival and expression of members of the Extended Myc Network. Based on RNAseq results from TCGA, the expression of each Extended Myc Network member was examined in the indicated tumors from the same database. Tumors were divided into subsets with overall favorable or unfavorable survival. The numbers in each of these subsets were based on the levels of transcript expression that provided the most significant survival differences. The “survival” and “survminer” R packages were used for survival analysis. FPKM value ranks were used to classify individuals into two groups by 50 series cutoffs ranging from 10 to 90%. Survivorship was examined by Kaplan–Meier survival estimators, and the survival outcomes of the two groups were compared by log-rank tests. Curves were based on the FPKM cutoffs that yielded maximal survival difference between the two groups with the lowest log-rank p-value. See Legend to Figure 2 for tumor type abbreviations.
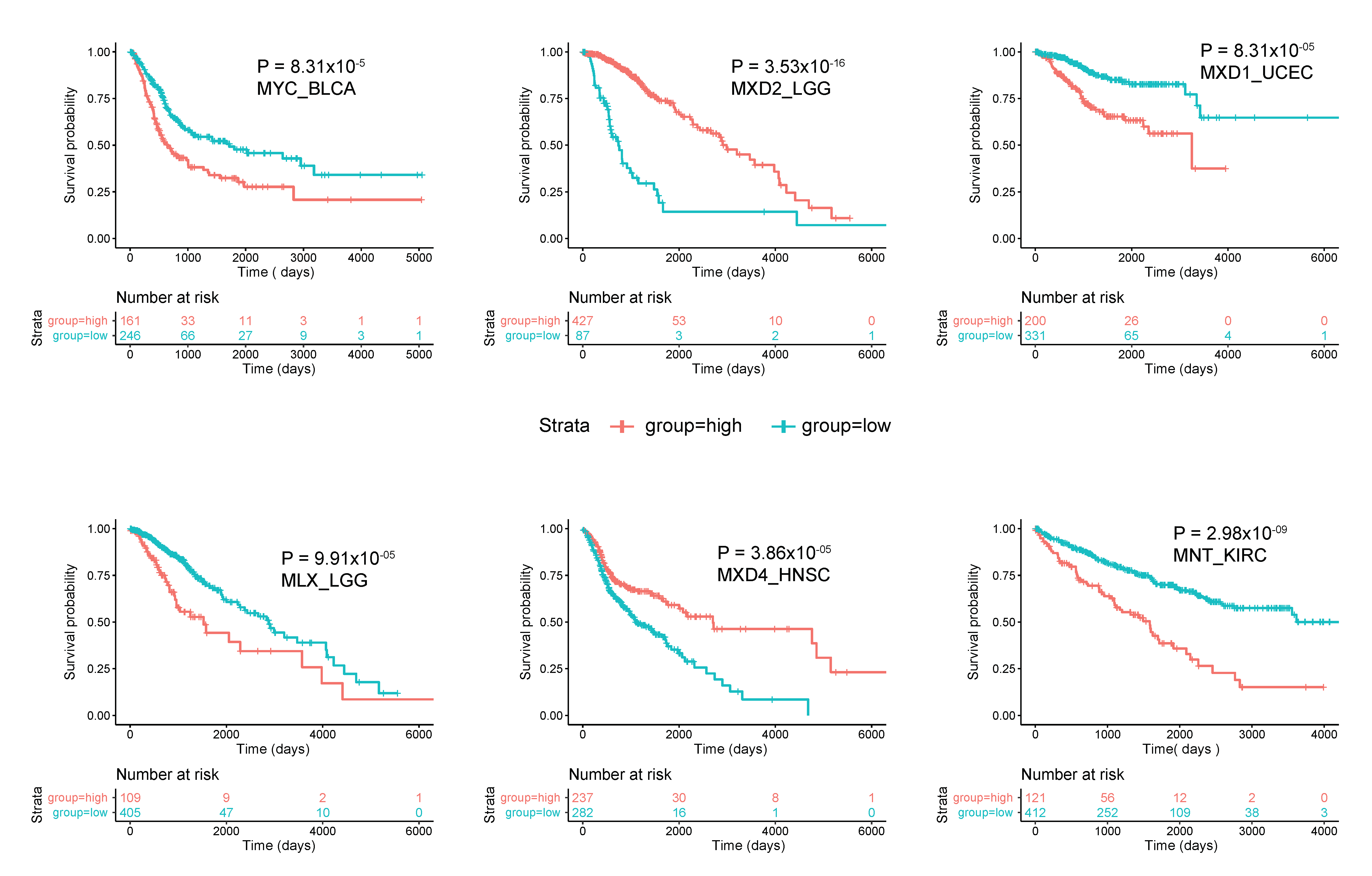
Figure 4. Correlations between survival and expression of select members of the Extended Myc Network. Tumors were chosen from those in TCGA and further subdivided as described in Figure 3. All curves were generated using the “survival” and “survminer” R packages. The number of individuals in the favorable and unfavorable survival groups which provided the greatest significance in survival is depicted in the “0 time point” beneath each survival curve.
The relationships among Extended Myc Network gene CNVs, mutation, expression and survival are in many cases only correlative. Indeed, this cautionary note applies even to an oncogene as unassailable as MYC whose role in tumorigenesis mandated a direct demonstration of its actual causality [113][114][115]. Yet, recent work has indicated that, at least for some tumor types, Myc may function more as a facilitator of tumor growth than as an actual initiator. For example, hepatoblastoma (HB) in mice, induced via the ectopic expression of mutant forms of β-catenin and the Hippo pathway effector protein YAP, is associated with Myc over-expression [78][116][117]. Yet, the rate of tumor induction in livers with hepatocyte-specific knockout of Myc is identical to that seen in wild-type livers [41][78] although tumor growth is slowed. Similarly, tumors induced with different β-catenin mutants express widely different levels of Myc that roughly correlate with growth rates even though they too are initiated at identical frequencies [117]. These results, strongly indicate that the role of Myc in actual tumor induction versus facilitation is quite nuanced and may reflect both the levels at which it is expressed and the tissue environment [118].
A somewhat analogous situation, discussed in greater detail below, occurs with Max. While there is no evidence that MAX is an actual oncogene, it could be viewed as being another tumor facilitator that collaborates with Myc (Figure 1). Yet, the recurrent loss of MAX in pheochromocytomas, parganglioneuromas and several other tumors, as shown in Figure 2 and elsewhere, supports its role as a potent TSG [119][120][121][122][123][124][125]. Similarly, MLX loss dramatically impairs normal hepatocyte proliferation while at the same time serving as a potent suppressor of benign hepatic adenomatosis [76].
2. Myc
Like RAS and PI3K, MYC is a classic example of a dominantly acting oncogene although, unlike the former two, it is seldom mutated in tumors. Rather, its oncogenicity commonly derives from gene amplification, where, along with CCND1 and EGFR, it is among the top three amplified genes across a broad range of cancers (Figure 2) [105]. Such frequent, pronounced and unidirectional changes in gene copy number emphasize this pro-oncogenic role and are rivaled, albeit only modestly so, by similar changes in CHREBP (Figure 2). In other tumors, such as Burkitt’s lymphoma and subsets of diffuse large B-cell lymphoma (DLBCL) and multiple myeloma, MYC’s over-expression is a result of its translocation into immunoglobulin gene loci [126][127][128][129]. Yet these examples, involving alterations in chromosomal architecture, underestimate the true frequency with which MYC is dysregulated in cancer as aberrant signaling by many mutant growth factor pathways converge on MYC and promote its over-expression in the absence of structural changes [130][131][132]. However, as mentioned in the Introduction, it is important to distinguish between MYC’s role as a primary driver oncogene versus that of a tumor facilitator that simply provides metabolic and/or translational support without being necessary for tumor initiation [41][78][113].
Unlike the direct induction of genes mediated by Myc–Max DNA binding mentioned above (Figure 1), Myc’s role in transcriptional suppression is indirect and involves the association of Myc–Max heterodimers with transcription factors such as Miz1 that normally up-regulate genes encoding negative growth regulators such as P15INK4B, P57KIP1 and P21CIP1 [133][134][135][136][137]. Mechanistically, Myc’s bHLH-ZIP domain binds directly to Miz1 and prevents the latter’s binding to initiator (Inr) elements at transcription initiation sites of these target genes. Similarly, Sp1’s binding to multiple GGGCGG sites located in the P21CIP1 promoter is disrupted via the interaction between its C-terminal zinc-finger and a ~200 residue internal region of Myc (amino acids 143–352) [138][139]. This effect is distinct from the suppression mediated by Miz1 since the deletion of P21CIP1’s Inr does not interfere with Myc’s blocking of SP1-mediated induction [138].
Despite its long and storied history as an oncoprotein, Myc may also play a role in tumor suppression. A novel means by which this might occur has been suggested by studies showing that ubiquitylation of several lysine residues in Myc’s C-terminus by the E3 ubiquitin ligase HectH9 is necessary for Myc to activate fully its target gene repertoire [140], possibly by licensing the latter protein’s interaction with and subsequent stabilization by the histone acetyltransferase (HAT) p300 [141]. HectH9 is over-expressed in many tumors and is inhibited by Miz1 [140]. This suggests that HectH9 is required for Myc to achieve its maximal transcriptional activation potential, presumably by optimizing its recruitment of p300 and other co-activators [16]. HectH9 thus represents a potential therapeutic target that could suppress if not entirely eliminate Myc’s impact on tumor growth by dictating its potency as an oncoprotein [140]. This further suggests that Myc toggles between two states of high and low transcriptional activation potential dictated by its degree of HectH9-mediated ubiquitination. Though perhaps not representing a true tumor suppressor in this latter context, the under ubiquitinated form of Myc could potentially behave in a dominant negative-like manner, thereby actively limiting tumor growth. Functionally, this could be viewed as being akin to one of the roles of Mxd proteins which is to functionally “inactivate” Myc by preventing its DNA binding (Figure 1).
Another post-translational modification that may contribute to Myc’s role in tumor suppression involves its direct phosphorylation at Thr358, S373 and Thr400 by the Pak2 Ser/Thr kinase, which inhibits Myc–Max heterodimerization and reduces target gene affinity [142]. In promyelocytic leukemia (PML) cells, this has an additional impact on retinoic acid (RA)-induced differentiation mediated by the RA receptor (RARα) and its induction of genes that inhibit proliferation and promote differentiation [143][144]. Unphosphorylated and phosphorylated Myc differentially contact RARα, with the former blocking RARα‘s transcriptional program and maintaining the proliferation of undifferentiated cells [145]. Unphosphorylated Myc’s binding to RARα decreases the latter’s association with co-activators while increasing its association with co-repressors, whereas the reverse occurs in response to Myc phosphorylation. Among the more prominent of these factors are the co-repressor HDAC3 histone deacetylase and the co-activator HAT CBP. The relationship between Myc and Pak2 is reciprocal in that Pak2 is up-regulated during late-stage RA-induced terminal PML differentiation when Myc levels are declining [146]. Thus, different environmental cues can allow Myc to either block or facilitate PML differentiation and proliferation. The mechanism by which Myc blocks PML differentiation, i.e., by directly interacting with and inhibiting another transcription factor, is formally analogous to the previous described inhibition by Myc of Miz1- and Sp1-mediated transcription.
Pak2-mediated phosphorylation of Myc has also been described in many other normal and neoplastic contexts, including primary hematopoietic cells, keratinocytes, fibroblasts and other leukemias [142][143][144][147][148][149]. It has been proposed that phosphorylated Myc plays a role in maintaining hematopoietic balance by promoting stem cell adherence and limiting expansion. However, the amplification of PAK2 (as well as PAK2-related genes) is considerably more common in cancer than is inactivation and missense mutations of Pak2 phosphorylation sites in Myc are virtually non-existent [143][144][150][151]. Thus, any role for Myc as a TS in primary human cancers is likely to be quite limited. The rare instances in which Myc seems to act as a “TS” are in actuality those in which post-translational modifications (or lack thereof) alter its stability or function in ways that reduce its effectiveness as an oncoprotein.
3. Max
Given Max’s central role in balancing the transcriptional output of the Myc Network (Figure 1), it might be perceived as promoting either pro-oncogenic or TS functions in a manner that depends on the relative abundance of Myc–Max heterodimers on the one hand and Mxd–Max heterodimers on the other [10][11][46][152]. Myc–Max association is needed to maintain both normal and tumor cell proliferation and inhibiting this for therapeutic purposes in cancer, whether by pharmacologic or genetic means, has been a long-desired goal [6][7][8][50][153][154][155][156][157][158]. Thus, disproportionate focus has been placed on Max in the context of its function as an obligatory pro-oncogenic co-factor for Myc-mediated transformation.
The first evidence for Max’s potential as a TS was provided by studies showing that, when co-expressed in relative excess to Myc, it suppressed Myc-mediated target gene activation and/or transformation in vitro [7][8][10][11][159][160]. Evidence for a natural TS function of Max was subsequently suggested by the observation that the PC12 pheochromocytoma (PC) cell line expressed a mutant Max transcript and no functional protein and that the enforced re-expression of wild-type Max inhibited PC12 growth [161].
Adrenal gland PCs and their extra-adrenal gland counterparts, paraganglioneuromas (PGLs), are typically benign tumors, approximately one-third of which are inherited and harbor identifiable germ-line mutations [162]. Additionally, approximately two-thirds of sporadic cases have an underlying genetic cause [119][163] involving at least 14 genes [120][121][122][125][164]. Max mutations are found in ~1–2% of tumors, particularly those which are inherited, display malignant features, arise in younger individuals and/or are associated with higher levels of metanephrine or normetanephrine secretion [120][122][162].
In the largest single study performed to date involving nearly 1700 inherited and sporadic PCs and PGLs, 20 Max mutations were identified, 18 of which were novel [120]. Nine of these were associated with mutations of the methionine initiation codon, with the remainder causing premature termination, aberrant splicing or in-frame deletion of crucial amino acids. The majority of these failed to express Max protein. The remaining 11 tumors contained missense mutations, seven of which were predicted to be deleterious, although all expressed the mutant Max proteins. Loss of heterozygosity (LOH) was observed in 16 of the 18 tumors analyzed.
MAX mutations have subsequently been described in tumors as diverse as multiple myeloma, small-cell lung cancer (SCLC), pituitary adenoma and quadruple wild-type gastrointestinal stromal tumor [165][166][167][168]. Multiple myeloma, in which the MAX mutation rate is ~3%, was a particularly informative source as the Max mutations provided clues into how they impacted the protein’s binding to various epigenetically-modified E-boxes [168]. The fact that methylation at position 5 of the internal cytosine residue (5mC) of the canonical CACGTG E-box inhibits Max binding has been known since the protein’s original discovery [169]. However, the E-box is subject to additional naturally-occurring cytosine modifications mediated by members of the ten-eleven translocation (Tet) family of Fe(II)- and α-ketoglutarate-dependent dioxygenase family, including 5-hydroxymethylation (5hmC), 5-formylation (5fC), and 5-carboxylation (5caC). All these modifications except 5caC have been shown to abrogate Max binding, particularly when the modification is present on both strands of the palindromic sequence [168]. A determination of Max’s crystal structure in homodimeric association with a 5caC-modified E-box showed that the R36 residue demonstrated the largest conformational difference when compared to the structure of Max or Myc–Max bound to the unmodified E-box. The importance of this residue was demonstrated by showing that the myeloma-associated Max mutation R36W completely abolished DNA binding. Moreover, of the 25 unique Max mutations identified among 805 multiple myelomas, 17 involved missense mutations, with five of these directly altering R36 and two altering the adjacent R35 residue. Finally, of the 643 tumor samples for which both mutational status and RNAseq results could be assessed, those harboring Max mutations expressed significantly lower levels of Myc and tended to display more favorable outcomes. The lower-level expression of Myc in these samples was likely due to the loss of its Max-mediated stabilization [168][170]. To explain how Myc drives myelomagenesis in the absence of efficient Max association, the authors suggested that Myc interacts with other E-box-binding proteins such as WDR5 [171][172].
SCLC in humans is often associated with TP53 and RB1 loss/mutation and amplification of MYC or its paralogs MYCN and MYCL [173][174][175]. A recent CRISPR/Cas9-based screen for suppressors of early-stage SCLC found Max to be among the top hits with no enrichment being observed for other members of the Extended Myc Network [176]. Confirmatory studies showed that Max knockdown in these “preSC” cells led to more robust growth and survival during both anchorage-dependent and independent growth in soft agar. When tested in vivo in an autochthonous Rb1/Trp53-deleted mouse model of SCLC [177], Max knockout was associated with significantly larger numbers of tumors and shortened survival. Max re-expression in a cell line derived from these tumor cells significantly suppressed growth. Interestingly, when preSC cells were engineered to over-express Myc, MycN or MycL, the concurrent knockout of Max inhibited growth. These findings suggested that, at least in this model system, Max’s role in SCLC development is context dependent and at least partly reliant upon the level of expression of Myc or its paralogs, particularly MYCL. While Max appeared to serve a pro-oncogenic function by facilitating MYC family-mediated transformation, it could also serve as a TS via one or more Myc-independent pathways.
RNAseq was performed in control preSC cells and SCLCs with or without MAX knockout as well as in SCLCs with MAX knockout and restored Max expression. Common deregulated genes shared by these cohorts included 113 that were up-regulated in response to MAX knockout and 56 that were down-regulated. Among the former were genes previously shown by ChIP to bind Myc–Max and Mxd–Max heterodimers. Collectively, these findings were consistent with the idea that MAX knockout reverses the suppression of genes mediated by the inhibitory arm of the Myc Network (Figure 1). Further analyses of these 113 genes and several hundred additional ones identified using less stringent criteria showed an enrichment for pathways dedicated to one-carbon metabolism along with the metabolism of nucleotide sugars, serine, alanine, aspartate and glutamate. Many of these genes’ promoters were co-occupied by Myc–Max and Mxd–Max heterodimers thus likely representing sites that could bind either heterodimer, depending on the cells’ metabolic and/or proliferative state. It was speculated that up-regulation of these genes in response to MAX knockout was due to the activity of other transcription factors that are otherwise normally impeded by Max–Mxd occupancy of nearby E-boxes. It was further surmised that the over-expression of MYC and its paralogs would displace Max–Mxd heterodimers from these sites, leading to increased Myc–Max binding and even more pronounced target gene induction.
The idea that Max plays a role in TS while also promoting transformation via its association with Myc suggests two simple and non-mutually exclusive ways by which this could occur. First, in some contexts, the loss of transcriptional balance between the positive and negative arms of the Myc Network may favor transformation due to distinct sets of genes whose expression is now altered (Figure 1). In other contexts, these target genes, no longer bound by any Myc Network components, are now excessively bound by other E-box-binding proteins and altered in ways the favor transformation.
References
- Alitalo, K.; Bishop, J.M.; Smith, D.H.; Chen, E.Y.; Colby, W.W.; Levinson, A.D. Nucleotide sequence to the v-myc oncogene of avian retrovirus MC29. Proc. Natl. Acad. Sci. USA 1983, 80, 100–104.
- Blackwood, E.M.; Eisenman, R.N. Max: A Helix-Loop-Helix Zipper Protein That Forms a Sequence-Specific DNA-Binding Complex with Myc. Science 1991, 251, 1211–1217.
- Reddy, E.P.; Reynolds, R.K.; Watson, D.K.; Schultz, R.A.; Lautenberger, J.; Papas, T.S. Nucleotide sequence analysis of the proviral genome of avian myelocytomatosis virus (MC29). Proc. Natl. Acad. Sci. USA 1983, 80, 2500–2504.
- Vennstrom, B.; Sheiness, D.; Zabielski, J.; Bishop, J.M. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J. Virol. 1982, 42, 773–779.
- Watson, D.K.; Reddy, E.P.; Duesberg, P.H.; Papas, T.S. Nucleotide sequence analysis of the chicken c-myc gene reveals homologous and unique coding regions by comparison with the transforming gene of avian myelocytomatosis virus MC29, delta gag-myc. Proc. Natl. Acad. Sci. USA 1983, 80, 2146–2150.
- Amati, B.; Dalton, S.; Brooks, M.W.; Littlewood, T.D.; Evan, G.I.; Land, H. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 1992, 359, 423–426.
- Amati, B.; Brooks, M.W.; Levy-Strumpf, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 1993, 72, 233–245.
- Amati, B.; Littlewood, T.; Evan, G.; Land, H. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 1993, 12, 5083–5087.
- Gu, W.; Cechova, K.; Tassi, V.; Dalla-Favera, R. Opposite regulation of gene transcription and cell proliferation by c-Myc and Max. Proc. Natl. Acad. Sci. USA 1993, 90, 2935–2939.
- Kretzner, L.; Blackwood, E.M.; Eisenman, R.N. Myc and Max proteins possess distinct transcriptional activities. Nature 1992, 359, 426–429.
- Kretzner, L.; Blackwood, E.M.; Eisenman, R.N. Transcriptional Activities of the Myc and Max Proteins in Mammalian Cells. Curr. Top. Microbiol. Immunol. 1992, 182, 435–443.
- Min, S.; Taparowsky, E.J. v-Myc, but not Max, possesses domains that function in both transcription activation and cellular transformation. Oncogene 1992, 7, 1531–1540.
- Kato, G.J.; Barrett, J.; Villa-Garcia, M.; Dang, C.V. An amino-terminal c-myc domain required for neoplastic transformation activates transcription. Mol. Cell. Biol. 1990, 10, 5914–5920.
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 2010, 29, 3691–3702.
- de Pretis, S.; Kress, T.R.; Morelli, M.J.; Sabò, A.; Locarno, C.; Verrecchia, A.; Doni, M.; Campaner, S.; Amati, B.; Pelizzola, M. Integrative analysis of RNA polymerase II and transcriptional dynamics upon MYC activation. Genome Res. 2017, 27, 1658–1664.
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 2021, 21, 579–591.
- Price, D.H. Regulation of RNA Polymerase II Elongation by c-Myc. Cell 2010, 141, 399–400.
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc Regulates Transcriptional Pause Release. Cell 2010, 141, 432–445.
- Berberich, S.J.; Cole, M.D. Casein kinase II inhibits the DNA-binding activity of Max homodimers but not Myc/Max heterodimers. Genes Dev. 1992, 6, 166–176.
- Prochownik, E.V.; VanAntwerp, M.E. Differential patterns of DNA binding by myc and max proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 960–964.
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-Interacting Protein Is Stimulated by Glucose through a Carbohydrate Response Element and Induces β-Cell Apoptosis. Endocrinology 2005, 146, 2397–2405.
- Hurlin, P.J.; Quéva, C.; Koskinen, P.; Steingrimsson, E.; Ayer, D.E.; Copeland, N.G.; Jenkins, N.A.; Eisenman, R.N. Mad3 and Mad4: Novel Max-interacting transcriptional repressors that suppress c-myc dependent transformation and are expressed during neural and epidermal differentiation. EMBO J. 1995, 14, 5646–5659.
- Popov, N.; Wahlström, T.; Hurlin, P.J.; Henriksson, M. Mnt transcriptional repressor is functionally regulated during cell cycle progression. Oncogene 2005, 24, 8326–8337.
- Quéva, C.; Hurlin, P.J.; Foley, K.P.; Eisenman, R.N. Sequential expression of the MAD family of transcriptional repressors during differentiation and development. Oncogene 1998, 16, 967–977.
- Yang, G.; Hurlin, P.J. MNT and Emerging Concepts of MNT-MYC Antagonism. Genes 2017, 8, 83.
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194.
- Zhou, Z.-Q.; Hurlin, P.J. The interplay between Mad and Myc in proliferation and differentiation. Trends Cell Biol. 2001, 11, S10–S14.
- Ayer, D.E.; Lawrence, Q.A.; Eisenman, R.N. Mad-max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell 1995, 80, 767–776.
- Llabata, P.; Mitsuishi, Y.; Choi, P.; Cai, D.; Francis, J.M.; Torres-Diz, M.; Udeshi, N.D.; Golomb, L.; Wu, Z.; Zhou, J.; et al. Multi-Omics Analysis Identifies MGA as a Negative Regulator of the MYC Pathway in Lung Adenocarcinoma. Mol. Cancer Res. 2020, 18, 574–584.
- Mathsyaraja, H.; Catchpole, J.; Freie, B.; Eastwood, E.; Babaeva, E.; Geuenich, M.; Cheng, P.F.; Ayers, J.; Yu, M.; Wu, N.; et al. Loss of MGA repression mediated by an atypical polycomb complex promotes tumor progression and invasiveness. eLife 2021, 10, e64212.
- Billin, A.N.; Ayer, D.E. The Mlx Network: Evidence for a Parallel Max-Like Transcriptional Network That Regulates Energy Metabolism. Curr. Top. Microbiol. Immunol. 2006, 302, 255–278.
- Chen, J.L.-Y.; Merl, D.; Peterson, C.W.; Wu, J.; Liu, P.Y.; Yin, H.; Muoio, D.M.; Ayer, D.E.; West, M.; Chi, J.-T. Lactic Acidosis Triggers Starvation Response with Paradoxical Induction of TXNIP through MondoA. PLoS Genet. 2010, 6, e1001093.
- Davies, M.N.; O’Callaghan, B.L.; Towle, H.C. Glucose Activates ChREBP by Increasing Its Rate of Nuclear Entry and Relieving Repression of Its Transcriptional Activity. J. Biol. Chem. 2008, 283, 24029–24038.
- Davies, M.N.; O’Callaghan, B.L.; Towle, H.C. Activation and repression of glucose-stimulated ChREBP requires the concerted action of multiple domains within the MondoA conserved region. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E665–E674.
- Li, M.V.; Chang, B.; Imamura, M.; Poungvarin, N.; Chan, L. Glucose-Dependent Transcriptional Regulation by an Evolutionarily Conserved Glucose-Sensing Module. Diabetes 2006, 55, 1179–1189.
- O’Shea, J.M.; Ayer, D.E. Coordination of Nutrient Availability and Utilization by MAX- and MLX-Centered Transcription Networks. Cold Spring Harb. Perspect. Med. 2013, 3, a014258.
- Peterson, C.W.; Stoltzman, C.A.; Sighinolfi, M.P.; Han, K.-S.; Ayer, D.E. Glucose Controls Nuclear Accumulation, Promoter Binding, and Transcriptional Activity of the MondoA-Mlx Heterodimer. Mol. Cell. Biol. 2010, 30, 2887–2895.
- Christopher, W.P.; Peterson, C.W.; Ayer, D.E. An extended Myc network contributes to glucose homeostasis in cancer and diabetes. Front. Biosci. 2011, 16, 2206–2223.
- Poungvarin, N.; Chang, B.; Imamura, M.; Chen, J.; Moolsuwan, K.; Sae-Lee, C.; Li, W.; Chanachai, S.-L. Genome-Wide Analysis of ChREBP Binding Sites on Male Mouse Liver and White Adipose Chromatin. Endocrinology 2015, 156, 1982–1994.
- Sans, C.L.; Satterwhite, D.J.; Stoltzman, C.A.; Breen, K.T.; Ayer, D.E. MondoA-Mlx Heterodimers Are Candidate Sensors of Cellular Energy Status: Mitochondrial Localization and Direct Regulation of Glycolysis. Mol. Cell. Biol. 2006, 26, 4863–4871.
- Wang, H.; Dolezal, J.M.; Kulkarni, S.; Lu, J.; Mandel, J.; Jackson, L.E.; Alencastro, F.; Duncan, A.W.; Prochownik, E.V. Myc and ChREBP transcription factors cooperatively regulate normal and neoplastic hepatocyte proliferation in mice. J. Biol. Chem. 2018, 293, 14740–14757.
- Wilde, B.; Ayer, D. Interactions between Myc and MondoA transcription factors in metabolism and tumourigenesis. Br. J. Cancer 2015, 113, 1529–1533.
- Yin, X.; Grove, L.; Prochownik, E.V. Lack of transcriptional repression by max homodimers. Oncogene 1998, 16, 2629–2637.
- Carroll, P.A.; Diolaiti, D. A novel role for the extended MYC network in cancer cell survival. Mol. Cell. Oncol. 2016, 3, e1026528.
- Diolaiti, D.; McFerrin, L.; Carroll, P.A.; Eisenman, R.N. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta 2015, 1849, 484–500.
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425.
- Poole, C.J.; Van Riggelen, J. MYC—Master Regulator of the Cancer Epigenome and Transcriptome. Genes 2017, 8, 142.
- Foley, K.P.; McArthur, G.A.; Quéva, C.; Hurlin, P.J.; Soriano, P.; Eisenman, R.N. Targeted disruption of the MYC antagonist MAD1 inhibits cell cycle exit during granulocyte differentiation. EMBO J. 1998, 17, 774–785.
- Foley, K.P.; Eisenman, R.N. Two MAD tails: What the recent knockouts of Mad1 and Mxi1 tell us about the MYC/MAX/MAD network. Biochim. Biophys. Acta 1999, 1423, M37–M47.
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad Network and the Transcriptional Control of Cell Behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699.
- Hurlin, P.J.; Quéva, C.; Eisenman, R.N. Mnt, a novel Max-interacting protein is coexpressed with Myc in proliferating cells and mediates repression at Myc binding sites. Genes Dev. 1997, 11, 44–58.
- Hurlin, P.J.; Steingrimsson, E.; Copeland, N.G.; Jenkins, N.A.; Eisenman, R.N. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. EMBO J. 2000, 19, 3841–3842.
- Hurlin, P.J.; Zhou, Z.; Toyo-Oka, K.; Ota, S.; Walker, W.L.; Hirotsune, S.; Wynshaw-Boris, A. Deletion of Mnt leads to disrupted cell cycle control and tumorigenesis. EMBO J. 2003, 22, 4584–4596.
- Hurlin, P.J.; Zhou, Z.-Q.; Toyooka, K.; Ota, S.; Walker, W.L.; Hirotsune, S.; Wynshaw-Boris, A. Evidence of mnt-myc antagonism revealed by mnt gene deletion. Cell Cycle 2004, 3, 95–97.
- Quéva, C.; McArthur, G.A.; Iritani, B.M.; Eisenman, R.N. Targeted Deletion of the S-Phase-Specific Myc Antagonist Mad3 Sensitizes Neuronal and Lymphoid Cells to Radiation-Induced Apoptosis. Mol. Cell. Biol. 2001, 21, 703–712.
- Billin, A.; Eilers, A.L.; Coulter, K.L.; Logan, J.S.; Ayer, D.E. MondoA, a Novel Basic Helix-Loop-Helix–Leucine Zipper Transcriptional Activator That Constitutes a Positive Branch of a Max-Like Network. Mol. Cell. Biol. 2000, 20, 8845–8854.
- Billin, A.N.; Eilers, A.L.; Queva, C.; Ayer, D.E. Mlx, a Novel Max-like BHLHZip Protein That Interacts with the Max Network of Transcription Factors. J. Biol. Chem. 1999, 274, 36344–36350.
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121.
- Han, K.-S.; Ayer, D.E. MondoA senses adenine nucleotides: Transcriptional induction of thioredoxin-interacting protein. Biochem. J. 2013, 453, 209–218.
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 2010, 395, 395–400.
- Petrie, J.L.; Al-Oanzi, Z.H.; Arden, C.; Tudhope, S.J.; Mann, J.; Kieswich, J.; Yaqoob, M.M.; Towle, H.C.; Agius, L. Glucose Induces Protein Targeting to Glycogen in Hepatocytes by Fructose 2,6-Bisphosphate-Mediated Recruitment of MondoA to the Promoter. Mol. Cell. Biol. 2013, 33, 725–738.
- Stoltzman, C.A.; Kaadige, M.R.; Peterson, C.W.; Ayer, D. MondoA Senses Non-glucose Sugars: Regulation of thioredoxin-interacting protein (txnip) and the hexose transport curb. J. Biol. Chem. 2011, 286, 38027–38034.
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by MondoA:Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917.
- Wilde, B.R.; Kaadige, M.R.; Guillen, K.P.; Butterfield, A.; Welm, B.E.; Ayer, D.E. Protein synthesis inhibitors stimulate MondoA transcriptional activity by driving an accumulation of glucose 6-phosphate. Cancer Metab. 2020, 8, 27.
- Wilde, B.R.; Ye, Z.; Lim, T.-Y.; Ayer, D.E. Cellular acidosis triggers human MondoA transcriptional activity by driving mitochondrial ATP production. eLife 2019, 8, e40199.
- Yu, F.-X.; Chai, T.F.; He, H.; Hagen, T.; Luo, Y. Thioredoxin-interacting Protein (Txnip) Gene Expression: Sensing oxidative phosphorylation status and glycolytic rate. J. Biol. Chem. 2010, 285, 25822–25830.
- Yu, F.-X.; Goh, S.-R.; Dai, R.-P.; Luo, Y. Adenosine-Containing Molecules Amplify Glucose Signaling and Enhance Txnip Expression. Mol. Endocrinol. 2009, 23, 932–942.
- Zhang, X.; Fu, T.; He, Q.; Gao, X.; Luo, Y. Glucose-6-Phosphate Upregulates Txnip Expression by Interacting with MondoA. Front. Mol. Biosci. 2020, 6, 147.
- Mejhert, N.; Kuruvilla, L.; Gabriel, K.R.; Elliott, S.D.; Guie, M.-A.; Wang, H.; Lai, Z.W.; Lane, E.A.; Christiano, R.; Danial, N.N.; et al. Partitioning of MLX-Family Transcription Factors to Lipid Droplets Regulates Metabolic Gene Expression. Mol. Cell 2020, 77, 1251–1264.e9.
- Carroll, P.; Diolaiti, D.; McFerrin, L.; Gu, H.; Djukovic, D.; Du, J.; Cheng, P.F.; Anderson, S.; Ulrich, M.; Hurley, J.B.; et al. Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis. Cancer Cell 2015, 27, 271–285.
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP•Mlx Is the Principal Mediator of Glucose-induced Gene Expression in the Liver. J. Biol. Chem. 2006, 281, 28721–28730.
- Ma, L.; Sham, Y.Y.; Walters, K.J.; Towle, H.C. A critical role for the loop region of the basic helix-loop-helix/leucine zipper protein Mlx in DNA binding and glucose-regulated transcription. Nucleic Acids Res. 2007, 35, 35–44.
- Ma, L.; Tsatsos, N.G.; Towle, H.C. Direct Role of ChREBP·Mlx in Regulating Hepatic Glucose-responsive Genes. J. Biol. Chem. 2005, 280, 12019–12027.
- Stoeckman, A.; Ma, L.; Towle, H.C. Mlx Is the Functional Heteromeric Partner of the Carbohydrate Response Element-binding Protein in Glucose Regulation of Lipogenic Enzyme Genes. J. Biol. Chem. 2004, 279, 15662–15669.
- Wutthisathapornchai, A.; Vongpipatana, T.; Muangsawat, S.; Boonsaen, T.; Macdonald, M.J.; Jitrapakdee, S. Multiple E-Boxes in the Distal Promoter of the Rat Pyruvate Carboxylase Gene Function as a Glucose-Responsive Element. PLoS ONE 2014, 9, e102730.
- Wang, H.; Lu, J.; Alencastro, F.; Roberts, A.; Fiedor, J.; Carroll, P.; Eisenman, R.N.; Ranganathan, S.; Torbenson, M.; Duncan, A.W.; et al. Coordinated Cross-Talk Between the Myc and Mlx Networks in Liver Regeneration and Neoplasia. Cell Mol. Gastroenterol. Hepatol. 2022; in press.
- Zhang, P.; Metukuri, M.R.; Bindom, S.M.; Prochownik, E.V.; O’Doherty, R.M.; Scott, D.K. c-Myc Is Required for the ChREBP-Dependent Activation of Glucose-Responsive Genes. Mol. Endocrinol. 2010, 24, 1274–1286.
- Wang, H.; Lu, J.; Edmunds, L.R.; Kulkarni, S.; Dolezal, J.; Tao, J.; Ranganathan, S.; Jackson, L.; Fromherz, M.; Beer-Stolz, D.; et al. Coordinated Activities of Multiple Myc-dependent and Myc-independent Biosynthetic Pathways in Hepatoblastoma. J. Biol. Chem. 2016, 291, 26241–26251.
- Denechaud, P.-D.; Bossard, P.; Lobaccaro, J.-M.A.; Millatt, L.; Staels, B.; Girard, J.; Postic, C. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. J. Clin. Investig. 2008, 118, 956–964.
- Denechaud, P.-D.; Dentin, R.; Girard, J.; Postic, C. Role of ChREBP in hepatic steatosis and insulin resistance. FEBS Lett. 2007, 582, 68–73.
- Havula, E.; Hietakangas, V. Glucose sensing by ChREBP/MondoA–Mlx transcription factors. Semin. Cell Dev. Biol. 2012, 23, 640–647.
- Havula, E.; Hietakangas, V. Sugar sensing by ChREBP/Mondo-Mlx—New insight into downstream regulatory networks and integration of nutrient-derived signals. Curr. Opin. Cell Biol. 2018, 51, 89–96.
- Havula, E.; Teesalu, M.; Hyötyläinen, T.; Seppälä, H.; Hasygar, K.; Auvinen, P.; Orešič, M.; Sandmann, T.; Hietakangas, V. Mondo/ChREBP-Mlx-Regulated Transcriptional Network Is Essential for Dietary Sugar Tolerance in Drosophila. PLoS Genet. 2013, 9, e1003438.
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. From The Cover: Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286.
- Jeong, Y.-S.; Kim, D.; Lee, Y.S.; Kim, H.-J.; Han, J.-Y.; Im, S.-S.; Chong, H.K.; Kwon, J.-K.; Cho, Y.-H.; Kim, W.K.; et al. Integrated Expression Profiling and Genome-Wide Analysis of ChREBP Targets Reveals the Dual Role for ChREBP in Glucose-Regulated Gene Expression. PLoS ONE 2011, 6, e22544.
- Ke, H.; Luan, Y.; Wu, S.; Zhu, Y.; Tong, X. The Role of Mondo Family Transcription Factors in Nutrient-Sensing and Obesity. Front. Endocrinol. 2021, 12, 653972.
- Lane, E.A.; Choi, D.W.; Garcia-Haro, L.; Levine, Z.G.; Tedoldi, M.; Walker, S.; Danial, N.N. HCF-1 Regulates De Novo Lipogenesis through a Nutrient-Sensitive Complex with ChREBP. Mol. Cell 2019, 75, 357–371.e7.
- Richards, P.; Ourabah, S.; Montagne, J.; Burnol, A.-F.; Postic, C.; Guilmeau, S. MondoA/ChREBP: The usual suspects of transcriptional glucose sensing; Implication in pathophysiology. Metabolism 2017, 70, 133–151.
- Richards, P.; Rachdi, L.; Oshima, M.; Marchetti, P.; Bugliani, M.; Armanet, M.; Postic, C.; Guilmeau, S.; Scharfmann, R. MondoA Is an Essential Glucose-Responsive Transcription Factor in Human Pancreatic β-Cells. Diabetes 2017, 67, 461–472.
- Airley, R.E.; McHugh, P.; Evans, A.R.; Harris, B.; Winchester, L.; Buffa, F.; Al-Tameemi, W.; Leek, R.; Harris, A. Role of carbohydrate response element-binding protein (ChREBP) in generating an aerobic metabolic phenotype and in breast cancer progression. Br. J. Cancer 2014, 110, 715–723.
- Buttgereit, F.; Brand, M. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995, 312 Pt 1, 163–167.
- Elgort, M.G.; O’Shea, J.M.; Jiang, Y.; Ayer, D.E. Transcriptional and Translational Downregulation of Thioredoxin Interacting Protein Is Required for Metabolic Reprogramming during G1. Genes Cancer 2010, 1, 893–907.
- Kaadige, M.R.; Looper, R.E.; Kamalanaadhan, S.; Ayer, D.E. Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating MondoA transcriptional activity. Proc. Natl. Acad. Sci. USA 2009, 106, 14878–14883.
- Kaadige, M.R.; Yang, J.; Wilde, B.R.; Ayer, D. MondoA-Mlx Transcriptional Activity Is Limited by mTOR-MondoA Interaction. Mol. Cell. Biol. 2015, 35, 101–110.
- Sipol, A.; Hameister, E.; Xue, B.; Hofstetter, J.; Barenboim, M.; Öllinger, R.; Jain, G.; Prexler, C.; Rubio, R.A.; Baldauf, M.C.; et al. MondoA Drives B-ALL Malignancy through Enhanced Adaptation to Metabolic Stress. Blood 2021, in press.
- Wernicke, C.M.; Richter, G.H.; Beinvogl, B.C.; Plehm, S.; Schlitter, A.M.; Bandapalli, O.R.; da Costa, O.P.; Hattenhorst, U.E.; Volkmer, I.; Staege, M.S.; et al. MondoA is highly overexpressed in acute lymphoblastic leukemia cells and modulates their metabolism, differentiation and survival. Leuk. Res. 2012, 36, 1185–1192.
- Afshar, A.R.; Pekmezci, M.; Bloomer, M.M.; Cadenas, N.J.; Stevers, M.; Banerjee, A.; Roy, R.; Olshen, A.B.; Van Ziffle, J.; Onodera, C.; et al. Next-Generation Sequencing of Retinoblastoma Identifies Pathogenic Alterations beyond RB1 Inactivation That Correlate with Aggressive Histopathologic Features. Ophthalmology 2020, 127, 804–813.
- Berry, J.L.; Polski, A.; Cavenee, W.K.; Dryja, T.P.; Murphree, A.L.; Gallie, B.L. The RB1 Story: Characterization and Cloning of the First Tumor Suppressor Gene. Genes 2019, 10, 879.
- Garber, J.E.; Offit, K. Hereditary Cancer Predisposition Syndromes. J. Clin. Oncol. 2005, 23, 276–292.
- Gargallo, P.; Yáñez, Y.; Segura, V.; Juan, A.; Torres, B.; Balaguer, J.; Oltra, S.; Castel, V.; Cañete, A. Li–Fraumeni syndrome heterogeneity. Clin. Transl. Oncol. 2020, 22, 978–988.
- Guha, T.; Malkin, D. Inherited TP53 Mutations and the Li–Fraumeni Syndrome. Cold Spring Harb. Perspect. Med. 2017, 7, a026187.
- Marmolejo, D.H.; Wong, M.Y.Z.; Bajalica-Lagercrantz, S.; Tischkowitz, M.; Balmaña, J.; Patócs, A.B.; Chappuis, P.; Colas, C.; Genuardi, M.; Haanpää, M.; et al. Overview of hereditary breast and ovarian cancer (HBOC) guidelines across Europe. Eur. J. Med Genet. 2021, 64, 104350.
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905.
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2.
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140.
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53.
- Kawate, S.; Fukusato, T.; Ohwada, S.; Watanuki, A.; Morishita, Y. Amplification of c-myc in Hepatocellular Carcinoma: Correlation with Clinicopathologic Features, Proliferative Activity and p53 Overexpression. Oncology 1999, 57, 157–163.
- Li, C.; Bonazzoli, E.; Bellone, S.; Choi, J.; Dong, W.; Menderes, G.; Altwerger, G.; Han, C.; Manzano, A.; Bianchi, A.; et al. Mutational landscape of primary, metastatic, and recurrent ovarian cancer reveals c-MYC gains as potential target for BET inhibitors. Proc. Natl. Acad. Sci. USA 2019, 116, 619–624.
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550.
- Singh, A.; Ham, J.; Po, J.; Niles, N.; Roberts, T.; Lee, C. The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy. Cells 2021, 10, 1082.
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41.
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849.
- Dolezal, J.M.; Wang, H.; Kulkarni, S.; Jackson, L.; Lu, J.; Ranganathan, S.; Goetzman, E.S.; Bharathi, S.S.; Beezhold, K.; Byersdorfer, C.A.; et al. Sequential adaptive changes in a c-Myc-driven model of hepatocellular carcinoma. J. Biol. Chem. 2017, 292, 10068–10086.
- Leder, A.; Pattengale, P.K.; Kuo, A.; Stewart, T.A.; Leder, P. Consequences of widespread deregulation of the c-myc gene in transgenic mice: Multiple neoplasms and normal development. Cell 1986, 45, 485–495.
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117.
- Tao, J.; Calvisi, D.F.; Ranganathan, S.; Cigliano, A.; Zhou, L.; Singh, S.; Jiang, L.; Fan, B.; Terracciano, L.; Armeanu–Ebinger, S.; et al. Activation of β-Catenin and Yap1 in Human Hepatoblastoma and Induction of Hepatocarcinogenesis in Mice. Gastroenterology 2014, 147, 690–701.
- Zhang, W.; Meyfeldt, J.; Wang, H.; Kulkarni, S.; Lu, J.; Mandel, J.A.; Marburger, B.; Liu, Y.; Gorka, J.E.; Ranganathan, S.; et al. β-Catenin mutations as determinants of hepatoblastoma phenotypes in mice. J. Biol. Chem. 2019, 294, 17524–17542.
- Soucek, L.; Evan, G.I. The ups and downs of Myc biology. Curr. Opin. Genet. Dev. 2010, 20, 91–95.
- Brito, J.P.; Asi, N.; Bancos, I.; Gionfriddo, M.R.; Zeballos-Palacios, C.L.; Leppin, A.L.; Undavalli, C.; Wang, Z.; Domecq, J.P.; Prustsky, G.; et al. Testing for germline mutations in sporadic pheochromocytoma/paraganglioma: A systematic review. Clin. Endocrinol. 2015, 82, 338–345.
- Burnichon, N.; Cascón, A.; Schiavi, F.; Morales, N.P.; Comino-Mendez, I.; Abermil, N.; Inglada, L.; de Cubas, A.A.; Amar, L.; Barontini, M.; et al. MAX Mutations Cause Hereditary and Sporadic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2012, 18, 2828–2837.
- Comino-Mendez, I.; Gracia-Aznárez, F.J.; Schiavi, F.; Landa, I.; Leandro-Garcia, L.; Letón, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667.
- Galan, S.R.; Kann, P.H. Genetics and molecular pathogenesis of pheochromocytoma and paraganglioma. Clin. Endocrinol. 2013, 78, 165–175.
- Korpershoek, E.; Koffy, D.; Eussen, B.H.; Oudijk, L.; Papathomas, T.G.; Van Nederveen, F.H.; Belt, E.J.T.; Franssen, G.J.H.; Restuccia, D.F.J.; Krol, N.M.G.; et al. Complex MAX Rearrangement in a Family With Malignant Pheochromocytoma, Renal Oncocytoma, and Erythrocytosis. J. Clin. Endocrinol. Metab. 2016, 101, 453–460.
- Roszko, K.L.; Blouch, E.; Blake, M.; Powers, J.; Tischler, A.; Hodin, R.; Sadow, P.; Lawson, E.A. Case Report of a Prolactinoma in a Patient With a Novel MAX Mutation and Bilateral Pheochromocytomas. J. Endocr. Soc. 2017, 1, 1401–1407.
- Welander, J.; Andreasson, A.; Juhlin, C.C.; Wiseman, R.W.; Bäckdahl, M.; Höög, A.; Larsson, C.; Gimm, O.; Söderkvist, P. Rare Germline Mutations Identified by Targeted Next-Generation Sequencing of Susceptibility Genes in Pheochromocytoma and Paraganglioma. J. Clin. Endocrinol. Metab. 2014, 99, E1352–E1360.
- Boxer, L.M.; Dang, C.V. Translocations involving c-myc and c-myc function. Oncogene 2001, 20, 5595–5610.
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306.
- Liu, Y.; Barta, S.K. Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2019, 94, 604–616.
- Schmitz, R.; Ceribelli, M.; Pittaluga, S.; Wright, G.; Staudt, L.M. Oncogenic Mechanisms in Burkitt Lymphoma. Cold Spring Harb. Perspect. Med. 2014, 4, a014282.
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137.
- Kaur, M.; Cole, M.D. MYC Acts via the PTEN Tumor Suppressor to Elicit Autoregulation and Genome-Wide Gene Repression by Activation of the Ezh2 Methyltransferase. Cancer Res. 2013, 73, 695–705.
- Wilkins, J.A.; Sansom, O.J. C-Myc Is a Critical Mediator of the Phenotypes of Apc Loss in the Intestine: Figure 1. Cancer Res. 2008, 68, 4963–4966.
- Gebhardt, A.; Frye, M.; Herold, S.; Benitah, S.A.; Braun, K.; Samans, B.; Watt, F.; Elsasser, H.-P.; Eilers, M. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J. Cell Biol. 2006, 172, 139–149.
- Seoane, J.; Pouponnot, C.; Staller, P.; Schader, M.; Eilers, M.; Massague, J. TGFβ influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat. Cell Biol. 2001, 3, 400–408.
- Si, J.; Yu, X.; Zhang, Y.; DeWille, J.W. Myc interacts with Max and Miz1 to repress C/EBPδ promoter activity and gene expression. Mol. Cancer 2010, 9, 92.
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Möröy, T.; Bartek, J.; Massague, J.; Hänel, F.; et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001, 3, 392–399.
- van Riggelen, J.; Müller, J.; Otto, T.; Beuger, V.; Yetil, A.; Choi, P.S.; Kosan, C.; Möröy, T.; Felsher, D.W.; Eilers, M. The interaction between Myc and Miz1 is required to antagonize TGFβ-dependent autocrine signaling during lymphoma formation and maintenance. Genes Dev. 2010, 24, 1281–1294.
- Gartel, A.L.; Shchors, K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp. Cell Res. 2003, 283, 17–21.
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515.
- Adhikary, S.; Marinoni, F.; Hock, A.; Hulleman, E.; Popov, N.; Beier, R.; Bernard, S.; Quarto, M.; Capra, M.; Goettig, S.; et al. The Ubiquitin Ligase HectH9 Regulates Transcriptional Activation by Myc and Is Essential for Tumor Cell Proliferation. Cell 2005, 123, 409–421.
- Faiola, F.; Liu, X.; Lo, S.; Pan, S.; Zhang, K.; Lymar, E.; Farina, A.; Martinez, E. Dual Regulation of c-Myc by p300 via Acetylation-Dependent Control of Myc Protein Turnover and Coactivation of Myc-Induced Transcription. Mol. Cell. Biol. 2005, 25, 10220–10234.
- Huang, Z.; Traugh, J.A.; Bishop, J.M. Negative Control of the Myc Protein by the Stress-Responsive Kinase Pak2. Mol. Cell. Biol. 2004, 24, 1582–1594.
- Uribesalgo, I.; Benitah, S.A.; Di Croce, L. From oncogene to tumor suppressor: The dual role of Myc in leukemia. Cell Cycle 2012, 11, 1757–1764.
- Uribesalgo, I.; Buschbeck, M.; Gutiérrez, A.; Teichmann, S.; Demajo, S.; Kuebler, B.; Nomdedeu, J.; Martín-Caballero, J.; Roma, G.; Benitah, S.A.; et al. E-box-independent regulation of transcription and differentiation by MYC. Nat. Cell Biol. 2011, 13, 1443–1449.
- Hörlein, A.J.; Näär, A.M.; Heinzel, T.; Torchia, J.; Gloss, B.; Kurokawa, R.; Ryan, A.; Kamei, Y.; Soderstrom, M.; Glass, C.K.; et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995, 377, 397–404.
- Nisimoto, Y.; Ogawa, H. Interaction between p21-activated protein kinase and Rac during differentiation of HL-60 human promyelocytic leukemia cell induced by all-trans-retinoic acid. Eur. J. Biochem. 2002, 269, 2622–2629.
- Benitah, S.A.; Frye, M.; Glogauer, M.; Watt, F.M. Stem Cell Depletion through Epidermal Deletion of Rac1. Science 2005, 309, 933–935.
- Huang, S.; Spector, D.L. Nascent pre-mRNA transcripts are associated with nuclear regions enriched in splicing factors. Genes Dev. 1991, 5, 2288–2302.
- Watt, F.M.; Frye, M.; Benitah, S.A. MYC in mammalian epidermis: How can an oncogene stimulate differentiation? Nat. Rev. Cancer 2008, 8, 234–242.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404.
- Chakraborty, A.A.; Scuoppo, C.; Dey, S.; Thomas, L.R.; Lorey, S.L.; Lowe, S.W.; Tansey, W.P. A common functional consequence of tumor-derived mutations within c-MYC. Oncogene 2015, 34, 2406–2409.
- Ayer, D.; Kretzner, L.; Eisenman, R.N. Mad: A heterodimeric partner for Max that antagonizes Myc transcriptional activity. Cell 1993, 72, 211–222.
- Amati, B.; Alevizopoulos, K.; Vlach, J. Myc and the cell cycle. Front. Biosci. 1998, 3, d250–d268.
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543.
- Jiang, H.; Bower, K.E.; Beuscher, A.E.; Zhou, B.; Bobkov, A.A.; Olson, A.J.; Vogt, P.K. Stabilizers of the Max Homodimer Identified in Virtual Ligand Screening Inhibit Myc Function. Mol. Pharmacol. 2009, 76, 491–502.
- Jung, K.-Y.; Wang, H.; Teriete, P.; Yap, J.L.; Chen, L.; Lanning, M.E.; Hu, A.; Lambert, L.J.; Holien, T.; Sundan, A.; et al. Perturbation of the c-Myc–Max Protein–Protein Interaction via Synthetic α-Helix Mimetics. J. Med. Chem. 2015, 58, 3002–3024.
- Prochownik, E.V.; Vogt, P.K. Therapeutic Targeting of Myc. Genes Cancer 2010, 1, 650–659.
- Whitfield, J.; Beaulieu, M.-E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10.
- Mukherjee, B.; Morgenbesser, S.D.; DePinho, R.A. Myc family oncoproteins function through a common pathway to transform normal cells in culture: Cross-interference by Max and trans-acting dominant mutants. Genes Dev. 1992, 6, 1480–1492.
- Prendergast, G.C.; Hopewell, R.; Gorham, B.J.; Ziff, E.B. Biphasic effect of Max on Myc cotransformation activity and dependence on amino- and carboxy-terminal Max functions. Genes Dev. 1992, 6, 2429–2439.
- Hopewell, R.; Ziff, E. The nerve growth factor-responsive PC12 cell line does not express the Myc dimerization partner Max. Mol. Cell. Biol. 1995, 15, 3470–3478.
- Jafri, M.; Maher, E.R. Genetics in Endocrinology: The genetics of phaeochromocytoma: Using clinical features to guide genetic testing. Eur. J. Endocrinol. 2012, 166, 151–158.
- Flynn, A.; Benn, D.; Clifton-Bligh, R.; Robinson, B.; Trainer, A.H.; James, P.; Hogg, A.; Waldeck, K.; George, J.; Li, J.; et al. The genomic landscape of phaeochromocytoma. J. Pathol. 2015, 236, 78–89.
- Maniam, P.; Zhou, K.; Lonergan, M.; Berg, J.N.; Goudie, D.R.; Newey, P.J. Pathogenicity and Penetrance of Germline SDHA Variants in Pheochromocytoma and Paraganglioma (PPGL). J. Endocr. Soc. 2018, 2, 806–816.
- Daly, A.F.; Castermans, E.; Oudijk, L.; Guitelman, M.A.; Beckers, P.; Potorac, I.; Neggers, S.J.C.M.M.; Sacre, N.; van der Lely, A.-J.; Bours, V.; et al. Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr.-Relat. Cancer 2018, 25, L37–L42.
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; De Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple WT GIST. Mol. Cancer Res. 2017, 15, 553–562.
- Romero, O.A.; Diz, M.T.; Pros, E.; Savola, S.; Gomez, A.; Moran, S.; Sáez, C.; Iwakawa, R.; Villanueva, A.; Montuenga, L.; et al. MAX Inactivation in Small Cell Lung Cancer Disrupts MYC–SWI/SNF Programs and Is Synthetic Lethal with BRG1. Cancer Discov. 2013, 4, 292–303.
- Wang, D.; Hashimoto, H.; Zhang, X.; Barwick, B.; Lonial, S.; Boise, L.; Vertino, P.M.; Cheng, X. MAX is an epigenetic sensor of 5-carboxylcytosine and is altered in multiple myeloma. Nucleic Acids Res. 2017, 45, 2396–2407.
- Prendergast, G.C.; Lawe, D.; Ziff, E. Association of Myn, the murine homolog of Max, with c-Myc stimulates methylation-sensitive DNA binding and ras cotransformation. Cell 1991, 65, 395–407.
- Mathsyaraja, H.; Freie, B.; Cheng, P.-F.; Babaeva, E.; Catchpole, J.T.; Janssens, D.; Henikoff, S.; Eisenman, R.N. Max deletion destabilizes MYC protein and abrogates Eµ-Myc lymphomagenesis. Genes Dev. 2019, 33, 1252–1264.
- Thomas, L.R.; Wang, Q.; Grieb, B.; Phan, J.; Foshage, A.M.; Sun, Q.; Olejniczak, E.T.; Clark, T.; Dey, S.; Lorey, S.; et al. Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol. Cell 2015, 58, 440–452.
- Thomas, L.R.; Adams, C.M.; Wang, J.; Weissmiller, A.M.; Creighton, J.; Lorey, S.L.; Liu, Q.; Fesik, S.W.; Eischen, C.M.; Tansey, W.P. Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad. Sci. USA 2019, 116, 25260–25268.
- Dammert, M.A.; Brägelmann, J.; Olsen, R.R.; Böhm, S.; Monhasery, N.; Whitney, C.P.; Chalishazar, M.D.; Tumbrink, H.L.; Guthrie, M.R.; Klein, S.; et al. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat. Commun. 2019, 10, 3485.
- McFadden, D.G.; Papagiannakopoulos, T.; Taylor-Weiner, A.; Stewart, C.; Carter, S.L.; Cibulskis, K.; Bhutkar, A.; McKenna, A.; Dooley, A.; Vernon, A.; et al. Genetic and Clonal Dissection of Murine Small Cell Lung Carcinoma Progression by Genome Sequencing. Cell 2014, 156, 1298–1311.
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285.
- Augert, A.; Mathsyaraja, H.; Ibrahim, A.H.; Freie, B.; Geuenich, M.J.; Cheng, P.-F.; Alibeckoff, S.P.; Wu, N.; Hiatt, J.B.; Basom, R.; et al. MAX Functions as a Tumor Suppressor and Rewires Metabolism in Small Cell Lung Cancer. Cancer Cell 2020, 38, 97–114.e7.
- Meuwissen, R.; Linn, S.C.; Linnoila, R.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
13 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No