Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria José Pérez Alvarez | -- | 3459 | 2022-04-10 19:18:21 | | | |
| 2 | Conner Chen | -10 word(s) | 3449 | 2022-04-11 02:35:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pérez Alvarez, M.J.; , . mTOR in the Brain under Physiological Conditions. Encyclopedia. Available online: https://encyclopedia.pub/entry/21550 (accessed on 24 July 2026).
Pérez Alvarez MJ, . mTOR in the Brain under Physiological Conditions. Encyclopedia. Available at: https://encyclopedia.pub/entry/21550. Accessed July 24, 2026.
Pérez Alvarez, Maria José, . "mTOR in the Brain under Physiological Conditions" Encyclopedia, https://encyclopedia.pub/entry/21550 (accessed July 24, 2026).
Pérez Alvarez, M.J., & , . (2022, April 10). mTOR in the Brain under Physiological Conditions. In Encyclopedia. https://encyclopedia.pub/entry/21550
Pérez Alvarez, Maria José and . "mTOR in the Brain under Physiological Conditions." Encyclopedia. Web. 10 April, 2022.
Copy Citation
ammalian/mechanistic target of rapamycin (mTOR) is a 289 kDa serine–threonine kinase and a key element of two mTOR complexes called mTORC1 and mTORC2 (mTORCs). Furthermore, mTOR is highly conserved and is the center of multiples signaling pathways and coordinates important cellular processes such as cell growth and metabolism. Although mTOR is ubiquitously expressed, it is especially abundant in the brain. Therefore, mTOR dysfunction profoundly affects the central nervous system (CNS).
mTOR
Hypoxia
Ischemia
Brain
Neuron
1. The Structure of mTOR and Its Complexes in the Brain
Mammalian/mechanistic target of rapamycin (mTOR) is a 289 kDa serine–threonine kinase and a key element of two mTOR complexes called mTORC1 and mTORC2 (mTORCs) [1][2][3][4]. Furthermore, mTOR is highly conserved and is the center of multiples signaling pathways and coordinates important cellular processes such as cell growth and metabolism [5]. Although mTOR is ubiquitously expressed, it is especially abundant in the brain [6]. Therefore, mTOR dysfunction profoundly affects the central nervous system (CNS). Mutations in genes encoding mTOR regulators induce neurological disorders called “mTORopathies” [5].
Furthermore, mTOR, as indicated by its name, is a target protein of rapamycin, an immunosuppressant and anti-fungal macrolide compound isolated from Streptomyces hygroscopicus. This kinase comprises several functional domains, including C-terminal small FAT domain (FATC), C-terminal kinase domain (KD), FKBP12 rapamycin-binding domain (FRB), transactivation/transformation-associated domain (FAT), and an N-terminal domain containing at least 20 HEAT (Huntingtin elongation factor 3 A subunit of PP2A TOR1) repeats. The latter provide sites for the interaction of regulatory proteins to form mTORC1 and mTORC2. The KD domain of mTOR, with conserved sequences homologous to the catalytic domain of the phosphoinositide 3-kinase (PI3K) family, contains phosphorylation sites that regulate the activity of this kinase [7].
Rapamycin and its analogs (called rapalogs) act as allosteric inhibitors of mTORC1 by interacting with the FRB domain of mTOR via FKBP12 protein (FK506-binding protein 1 A 12 kDa) [6]. Furthermore, mTORC2 is insensitive to rapamycin inhibition, as initially described [8], but prolonged exposure to this macrolide results in the disruption of the assembly and integrity of mTORC2, thereby causing the functional inhibition of the complex [9].
Additionally, mTORC1 and mTORC2 share several common proteins, including the catalytic subunit mTOR, Deptor (DEP-domain-containing mTOR interacting protein), mLST8 (mammalian lethal with Sec13 protein 8), and Tti1/Tel2 complex [10] (Figure 1). In addition, each complex has specific proteins. Raptor (regulatory-associated protein of mTOR) and PRAS40 (proline-rich Akt substrate 40 kDa) are specific subunits of mTORC1, while Rictor (rapamycin-insensitive companion of mTOR), mSin1, and Protor1/2 are exclusive to mTORC2 [7] (Figure 1). All of these proteins have different functions in the complexes. Not only do they have structural functions (stabilizing the complexes and recruiting mTOR substrates) but they also contribute to regulating mTOR activity. Recently, an advancement in understanding of the precise functions of each mTOR companion protein beyond the strictly structural function has occurred. Recent studies demonstrate an important impact in the fine-tuned activity of mTOR kinase, according to post-translational modifications of some of the companion proteins, mainly by phosphorylation.
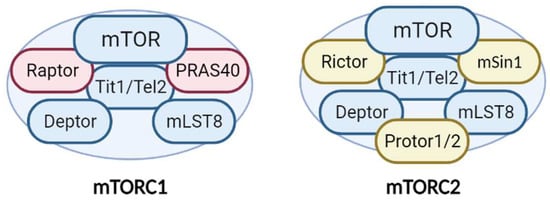
Figure 1. Structure and components of the mTORC1 and mTORC2 complexes. The mTORCs share common proteins (blue) called mTOR (catalytic subunit), Deptor (mTOR inhibitor subunit), mLST8 (scaffold and activator subunit), and Tit/Tel2 (assembly subunit). The specific mTORC1 proteins (pink) include Raptor and PRAS40 (mTOR inhibitor). Rictor, mSIN1, and Protor1/2 (activity modulator) comprise mTORC2 (yellow). See text for more details.
Deptor is an inhibitor of mTOR. The ablation of Deptor increases tumor size and causes the proliferation, migration, and invasion of tumoral cells because of the resulting overactivation of mTOR [11]. Furthermore, several post-translational modifications of Deptor can affect its inhibitory function. Recent studies show that the phosphorylation of Deptor at Tyr289 increases mTOR activity by preventing the correct coupling of the kinase to its protein-binding partner to form a complex [12][13]. This observation reveals the involvement of a novel molecular switch in the fine regulation of mTORCs.
The importance of mLST8 in mTORC activity is under debate. Studies using Drosophila melanogaster reveal that mLST8 is essential for mTORC2 activity, since LST8 knockout conserves mTORC1 but not mTORC2 activity [14]. It has been proposed that mLST8 specifically affects the interaction between mTOR and Rictor and is essential for the proper assembly of mTORC2 [15]. However, a recent study supports the notion that mLST8 is equally essential for ensuring the stability and activation of the two complexes through a mechanism that involves the kinase Akt [16]. PRAS40 is a negative regulator of mTORC1. Akt phosphorylation of PRAS40 increases the inhibitory effect on mTORC1, measured as an increase in autophagy [17]. Furthermore, mSin1 has been considered a scaffold protein with no relevance for mTOR activity [18]. However, recent data suggest that mSin1 is more than a structural protein. Indeed, it is essential for Akt phosphorylation at Ser473 by mTORC2 [19]. In addition, mSin1 defines the subcellular location of mTORC2, which also determines the final activity of the kinase. Alternative splicing of mSin1 generates five isoforms, of which at least three are able to assemble into mTORC2 and which determine three distinct mTORC2 complexes. These can be located in different subcellular compartments and their sensitivity to activation by PI3K differs [19].
From a functional perspective, mTORCsare considered molecular sensors of cellular energy status. They monitor several extra- and intra-cellular factors to orchestrate a response to maintain cellular homeostasis [20]. Furthermore, mTORC1 is involved in key cellular anabolic and catabolic functions, such as the synthesis of macromolecules (both lipids and proteins), cellular growth, autophagy, and cell cycle progression (Figure 2). In contrast, mTORC2 is related mainly to cell survival, as well as cytoskeletal organization and remodeling [21] (Figure 2). Therefore, both mTORCs are essential for cell viability, as evidenced using knockout mice for mTOR, Raptor, or Rictor. In all of these mouse models, embryo viability is severely compromised [22][23][24][25].
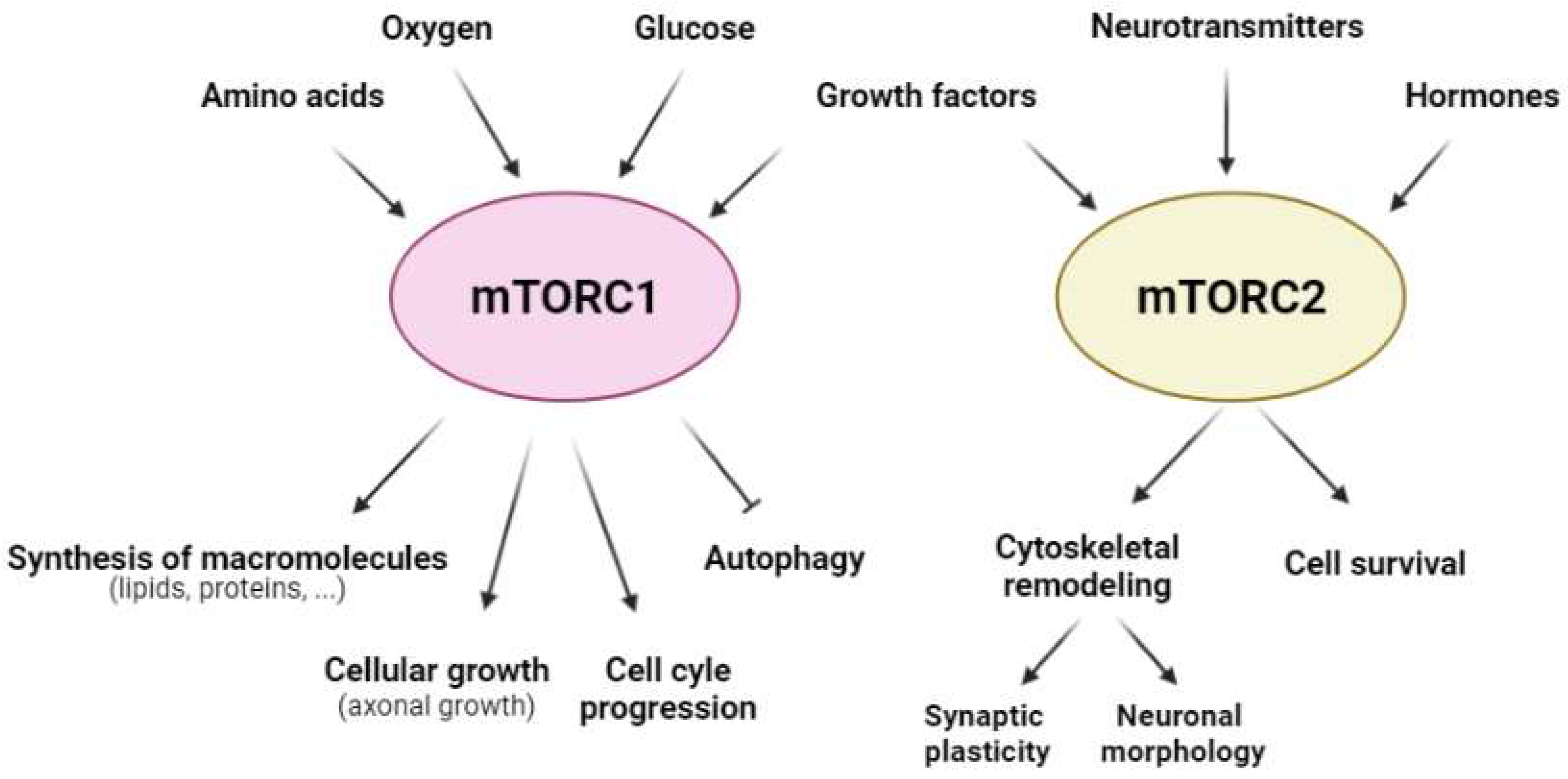
Figure 2. Regulatory factors of mTORC1 and mTORC2 and the cellular responses that they govern. The activity of mTORC1 (pink) and mTORC2 (yellow) is modulated by a range of extra- and intra-cellular factors; mTORC1 (pink) is regulated mainly by amino acids, oxygen, glucose, and growth factors, whereas mTORC2 (yellow) is dependent on growth factors, neurotransmitters, and hormones in the CNS. Furthermore, mTORC1 regulates anabolic and catabolic cellular processes, such as the synthesis of macromolecules (lipids and proteins), cellular growth, cell cycle progression, and autophagy; mTORC2 modulates cellular processes that involve cytoskeletal remodeling, such as synaptic plasticity and neuronal morphology, as well as cell survival.
As both mTORC1 and mTORC2 regulate important cellular functions, their activity is finely regulated. Knowledge of the regulation of mTORC1 is currently much greater than that of mTORC2. Given that the latter is an important player in the maintenance of cell survival and may be an interesting target in neurodegenerative diseases, it consider that greater efforts should be devoted to unraveling the fine details of mTORC2 regulation.
The principal mechanism of mTOR activation are related to post-translational modifications (mainly phosphorylation). The mTORCs have multiple regulatory phosphorylation sites, not only in the catalytic subunit (mTOR), but also in other subunits of the complex, such as Raptor, Rictor, Deptor, and PRAS40 [26][27][28]. The data available point to a relationship between the degree of phosphorylation of each subunit and the activity levels of mTOR. This notion opens up a new perspective of the fine regulation of mTOR activity, which could be modulated similarly to volume control and would differentially affect the phosphorylation levels of the substrates and their activities. It is important to keep this perspective in mind in the context of brain ischemia, since the energy status of cells changes rapidly and heterogeneously depending on the degree of injury [29].
2. Upstream Regulatory Pathways of mTORCs
One of the most remarkable characteristics of mTORCs, especially mTORC1, is that these complexes are cellular energy sensors, and as such they act as signal convergence centers from extra- and intra-cellular “energetic factors”. The presence or absence of these factors modulates the final activity of mTORC1, allowing it to trigger distinct cellular responses to modify the balance between anabolism and catabolism, adjusting it to cellular needs (Figure 3).
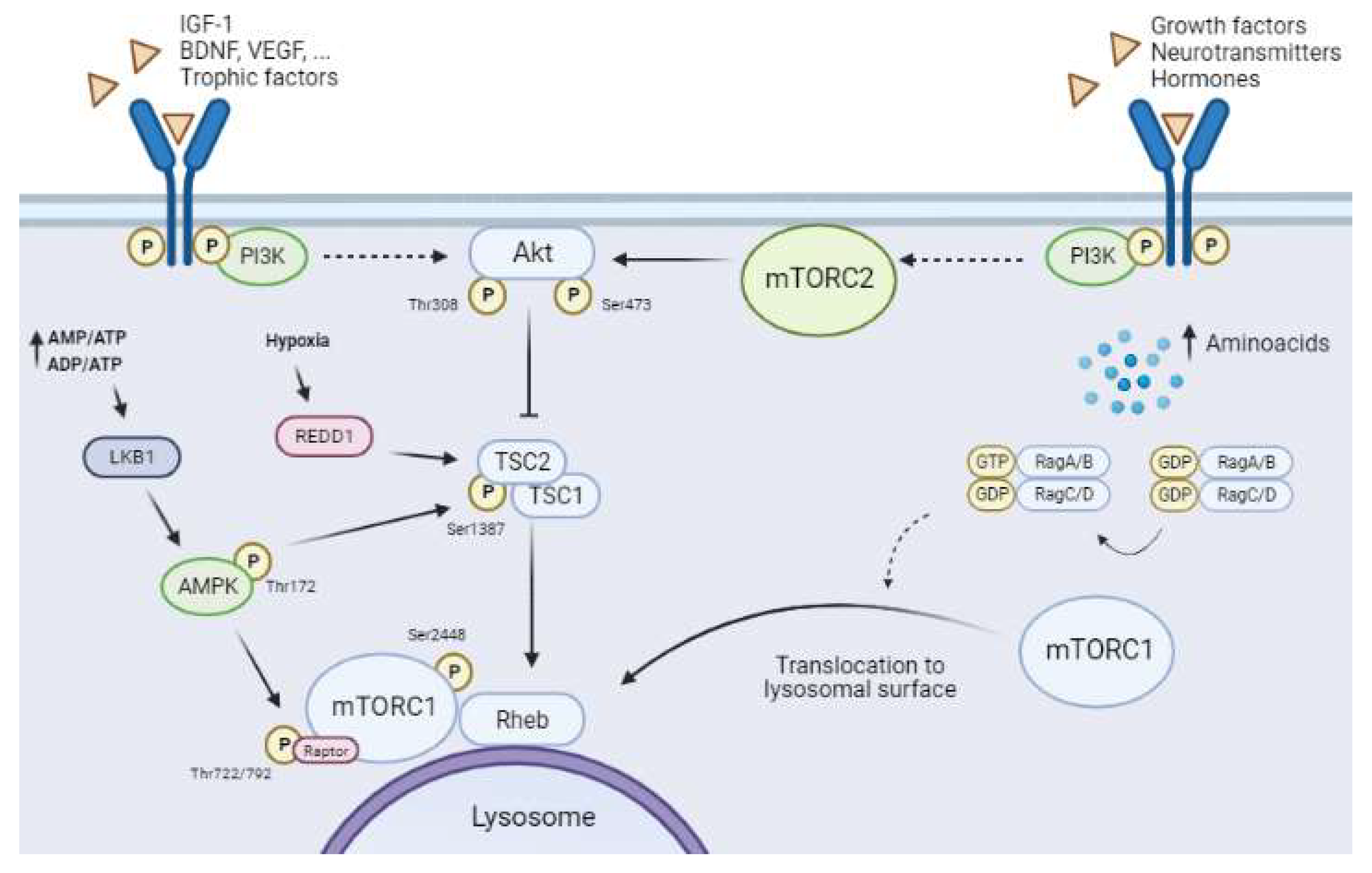
Figure 3. Upstream mTORCs pathways. Solid arrows show direct interactions between proteins and dashed arrows indicate the presence of other unrepresented mediators. The canonical PI3K/Akt pathway regulates mTORC1 activity through the binding of trophic and growth factors (BDNF, VEGF, IGF-1 among others) to RTK/GPCR-specific receptors. PI3K activation upregulates Akt by phosphorylation of Thr308. Full Akt activity requires the phosphorylation of Ser473 by mTORC2. Active Akt inhibits the tuberous sclerosis complex (TSC), which leads to induction of the GTPase Rheb, allowing activation of mTORC1. Amino acid availability leads to the translocation of mTORC1 to the lysosome membrane, which in turn allows its activation by Rheb. The AMPK pathway is activated in low-energetic states (AMP/ATP and ADP/ATP ratios increase). This activation requires the phosphorylation of Thr172 by LKB1. Active AMPK reduces mTORC1 activity through two distinct mechanisms, namely phosphorylation of Raptor at Ser722/792 and phosphorylation of TSC2 at Ser1387. Hypoxia induces an increase in REDD1, which downregulates mTORC1 by destabilizing TSC. Furthermore, mTORC2 activity is regulated by the presence of growth factors, neurotransmitters, and hormones, which all activate PI3K by binding to RTK/GPCRs.
Generally, the kinase activity of mTORCs promotes cellular anabolic pathways (translation, transcription, and lipid synthesis) and downregulates catabolism (protein degradation and autophagy). Interestingly, both complexes show a hierarchical position in the signaling pathways and are regulated by specific extra- and intra-cellular factors.
Furthermore, mTORC1 is regulated by multiple pathways related to the availability of trophic and growth factors, nutrients (especially amino acids and glucose), and oxygen (Figure 3). In contrast, mTORC2 activity is regulated mainly by growth factors, hormones, and neurotransmitters by the upregulation of PI3K activity [5][30]. However, knowledge about the regulation of mTORC2 in the CNS is scarce. Several studies show the importance of mTORC2 in cytoskeletal organization and remodeling and neuronal morphology. Neuronal mTORC2 is activated by neurotrophins, glutamate, NMDA, and inducers of long-term potentiation (LTP), the latter being a pivotal mechanism in synaptic plasticity and memory [30].
2.1. The Canonical Pathway: PI3K/Akt/mTORC1 and Growth Factors
This pathway senses the availability of several growth or survival factors, including nerve growth factor (NGF), brain-derived growth factor (BDNF), vascular endothelial growth factor (VEGF), insulin, insulin-like growth factor-1 (IGF-1), and neurotrophins (NT-1, -3, and -4). These molecules activate the PI3K/protein kinase B (PI3K/Akt) pathway by binding to their specific membrane receptors, which belong to the tyrosine kinase receptor superfamily (RTK), or to G-protein-coupled receptors (GPCRs) [20]. Full Akt activation requires its phosphorylation at two sites, namely Thr308 via the PI3K/Akt pathway and Ser473 via mTORC2 activity (Figure 3). First, Akt must be recruited to the cellular plasma membrane by direct interaction with phosphatidylinositol (3,4,5)-triphosphate (PIP3) [31]. The Ser473 phosphorylation site reveals the exquisite relationship between the activities of the two mTORCs, as mTORC1 activation requires full Akt activity, which in turn calls for previous mTORC2 activation [32]. Some studies show that Akt activity is finely regulated and that this protein kinase requires phosphorylation at other residues, such as Ser477 and Thr479, to enhance its interaction with mTORC2 and stability [31].
Akt activation induces the inhibition of TSC (Figure 3), a trimeric complex formed by TSC1 (hamartin), TSC2 (tuberin), and the scaffold protein TSC1D7. Several pathways converge on TSC to regulate (both positively and negatively) mTORC1 activity [27]. TSC is a GTPase-activating protein for the small GTPase Rheb (Ras homolog enriched in brain). Rheb is present in an inactive or activated state by binding to GDP or GTP, respectively [33]. Akt phosphorylates TSC2 at Thr1462, thereby disassembling and inactivating the complex. This process allows a Rheb-GTP-active state, which consequently induces mTORC1 through an unknown mechanism (Figure 3) [33][34]. The subcellular localization of mTORC1 in this step is essential for its full activation, as previously mentioned, since Rheb-GTP is located in the lysosomal membrane (Figure 3).
2.2. AMPK–mTORC1 Pathway: The Glucose Sensor
AMPK, a negative regulator of mTORC1 activity, comprises three subunits (α, β, and γ), and it senses cellular energy status and glucose availability [35]. The highly energy-demanding brain is unable to store glucose and is greatly dependent on constant glucose supply from blood. A decrease in glucose supply induces a reduction in the AMP/ATP and ADP/ATP ratios, which is sensed by AMPK (Figure 3). This detection induces AMPK activation by phosphorylation at Thr172 of its α-subunit by LKB1 tumor suppression kinase [36]. Ca2+-activated Ca2+/calmodulin-dependent kinase β (CaMKKβ) and transforming growth factor-β-activating kinase 1 (TAK1) are both activators of AMPK [37][38].
The activation of AMPK inhibits mTORC1 activity through phosphorylation on two targets, namely TSC2 and Raptor [39]. Therefore, AMPK regulates mTORC1 activity at two levels (Figure 3). On the one hand, AMPK phosphorylation of TSC2 at Thr1227 and Ser1345 improves the stability of the TSC complex [33], promoting the inactive state of Rheb-GDP and inducing the inhibition of mTORC1 activity [40]. On the other hand, Raptor phosphorylation at Ser722/792 by AMPK disrupts mTORC1 and induces its inhibition [41]. AMPK activation and mTORC1 inhibition lead to autophagy.
A reduction in oxygen levels, as occurs under hypoxia, decreases cellular ATP levels by inhibiting oxidative phosphorylation and other metabolic programs. This scenario promotes an ATP/AMP imbalance, inducing AMPK activation [42] and mTORC1 inhibition.
2.3. REDD1 and mTORC1
Reduced oxygen availability is another scenario that negatively modulates mTORC1 [43] (Figure 3). Hypoxia induces an increase in regulated DNA damage and development 1 (REDD1; also known as RTP801/DDIT4) expression, a highly conserved stress-response protein [44]. REDD1 downregulates mTORC1 through a TSC-dependent mechanism [45][46]. REDD1 triggers the release of TSC2 with the adapter protein 14-3-3, stabilizing the interaction between TSC1 and TSC2 and inducing mTORC1 inhibition [45]. Increased REDD1 expression early after ischemia has been described in neurons and glial cells [47].
2.4. Regulation of mTORC1 by Amino Acid Levels
Amino acids are key elements for neural cells. Moreover, amino acids are another regulatory pathway of mTORC1 involving a molecular mechanism that is not fully understood. Amino acids levels have an important influence on mTORC1 activity as they mediate the translocation of these kinases to the lysosomal membrane, a necessary step for it activation [21][34]. Rag GTPases have been reported to be involved in this process [21][34][48][49]. These molecules are small G-proteins that belong to the Ras superfamily andare present as heterodimers—RagA/B dimerized with RagC/D. The active conformation of these Rag heterodimers is RagA/B binding to GTP (RAG-A/BGTP) and RagC/D binding to GDP (RAG-C/DGDP) (Figure 3) [50]. Amino acid availability allows the active conformation of Rag, which binds directly to Raptor and induces the recruitment of mTORC1 to the lysosomal membrane [51] (Figure 3). At this location, mTORC1 is accessible to Rheb as a result of their proximity, and consequently mTORC1 is activated [52][51]. Leucine is a key amino acid that influences mTORC1 activity as it enhances the stabilization of the Raptor–mTOR interaction [53].
Cerebral ischemia is followed by a reduction in blood flow to the brain. This disruption causes the dysregulation of all upstream pathways that regulate mTORC1 . However, the impact of each individual upstream pathway on the activity of this complex is unknown. In this regard, it would be interesting to determine the relevance of each pathway for the final activity of mTORC1, which could differ between cell types. Preliminary data obtained in laboratory using primary cultures of neurons and astrocytes suggest that the reductions in several energetic factors (glucose, oxygen, and trophic factors) have a summative effect on the activity levels of mTORC1.
3. Downstream Targets of mTORCs
The strategic position of mTORC1, downstream of three important signaling pathways, makes this complex an essential convergence center to check cell energy status. Here describe the signaling pathways downstream of mTORC1, their principal targets, and the main cellular processes that they regulate. In addition, here provide the scarce details available for the downstream pathways of mTORC2 in the CNS.
One of the best known cellular processes regulated by mTORC1 is protein synthesis (Figure 4), which is essential for neural cell survival, synaptic plasticity, and brain development, and is dysregulated in several brain conditions, such as ischemia [27]. In neurons, trophic factors such as BDNF, insulin, and IGF-1, as well as some neurotransmitters, induce an increase in protein synthesis by local mTORC1 activation [54][55]. Protein synthesis is regulated by two well-characterized targets of mTORC1, called p70 ribosomal protein S6 kinase (P70S6K) and eukaryotic initiation factor 4E (eIF4E)-binding proteins (4EBPs), which trigger elongation and initiation of protein translation, respectively [56] (Figure 4). Furthermore, mTORC1 phosphorylates 4EBPs at several residues, thereby allowing the release of a group of eukaryotic initiation factors (eIFs) located on 5’-UTR of cap-dependent mRNA that initiates translation [57][58]. Three isoforms of 4EBP have been described, namely 4EBP-1, 4EBP-2, and 4EBP-3, with 4EBP-2 being the most common in the CNS [58]. Additionally, mTORC1 phosphorylates 4EBPs mainly at Thr37/46 and Ser65. It has been proposed that hierarchical phosphorylation of 4EBPs may be crucial in the fine regulation of translational processes mediated by mTORC1 [59][60][61]. P70S6K is the other main substrate of mTORC1 related to protein synthesis. Additionally, mTORC1 phosphorylates P70S6K at Thr389, thereby allowing the recruitment of the 40S ribosomal subunit to the translational machinery. Furthermore, P70S6K phosphorylates eukaryotic initiation Factor 2 kinase (eIF2K), which induces the elongation phase of protein synthesis [21].
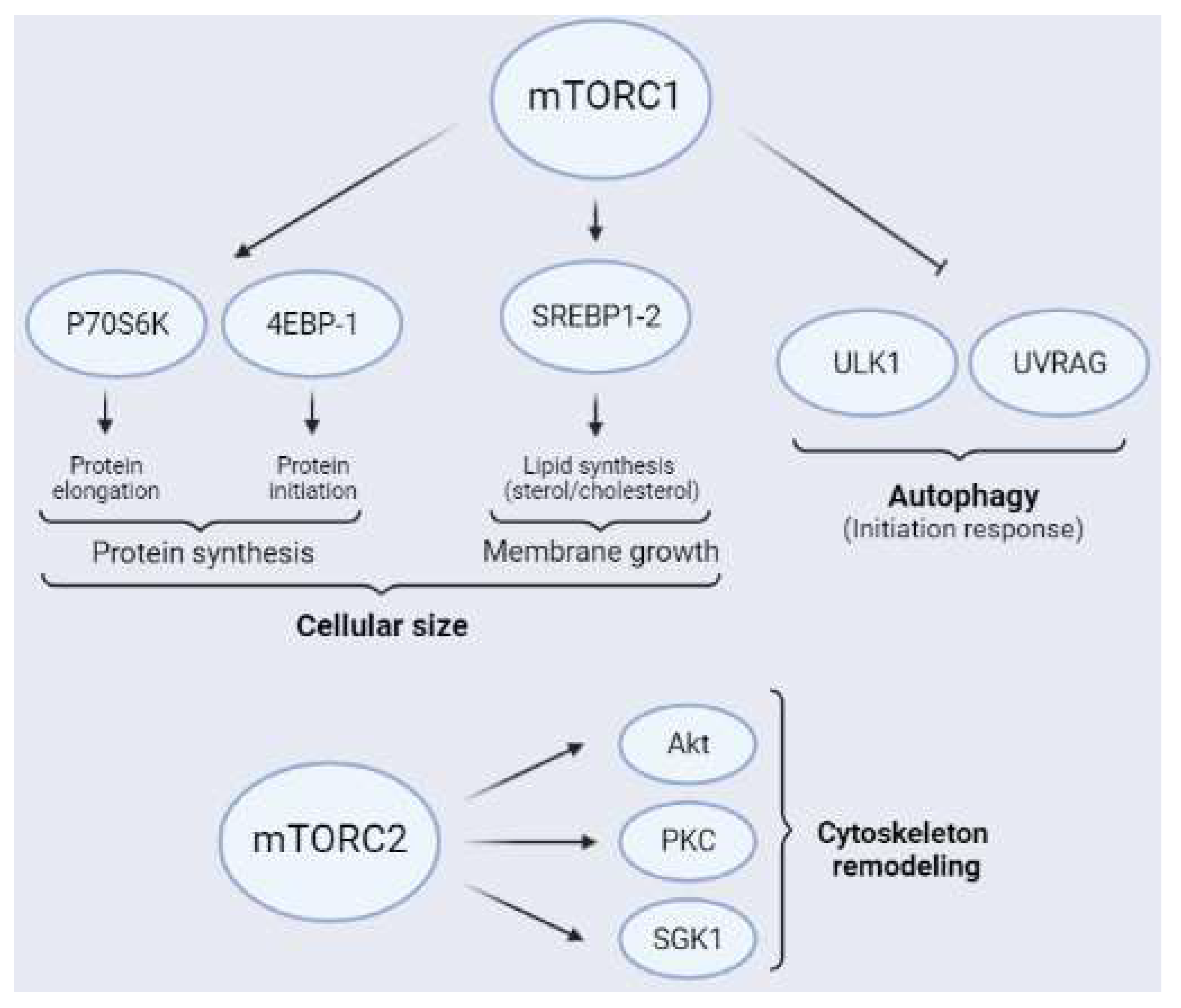
Figure 4. Schematic representation of the main mTORC1 (above) and mTORC2 (below) substrates. The main targets of mTORC1 related to protein synthesis are P70S6K and 4EBPs. Furthermore, mTORC1 regulates lipid synthesis through the transcriptional factor of lipogenesis SREBP1-2 and inhibits autophagy through ULK1 and UVRAG, the latter mainly in neurons. Additionally, mTORC2 is an activity modulator of Akt, PKC, and SGK1, all of which are related to cytoskeleton remodeling.
The synthesis of structural lipids allows the length of the plasmatic membrane to increase, a key aspect in axonal growth, dendritic arborization, and myelination (Figure 4). In addition, lipids are important molecules in cellular metabolic processes, since they are sources of energy, especially when glucose is lacking, as occurs after ischemia [62][63][64]. Furthermore, mTORC1 inhibition using rapamycin downregulates lipid synthesis. This observation, reveals that mTORC1 participates in this process [65]. Additionally, mTORC1 activates sterol regulatory element-binding proteins 1 and 2 (SREBP1-2) through P70S6K, since ablation of P70S6K induces reductions in the amount of lipid synthesis and cell size [66]. Activated SREBP is translocated to the nucleus to act as a transcription factor of genes involved in lipogenesis [67][68]. In the developing brain, mTORC1 induces the transcription of numerous genes related to the sterol–cholesterol biosynthesis pathway. Altered expression of these genes by dysregulation of mTORC1 contributes to neurodevelopmental disorders [69] and also affects myelination [70], an essential process for neuronal recovery after cerebral ischemia.
Autophagy is a highly conserved cellular catabolic mechanism that involves the degradation of damaged cellular components, misfolded proteins, long-lived proteins, and damaged organelles by lysosomes. It is believed that after injury, autophagy plays a critical role in removing damaged molecules and subcellular components to maintain cellular homeostasis [71]. Furthermore, mTORC1 is a key player in the regulation of autophagy (Figure 4). It induces this catabolic mechanism and facilitates the fusion of the autophagosome with the lysosome, a key step in this process [72]. In nutrient-rich conditions, active mTORC1 inhibits autophagy by phosphorylating Unc-51-like kinase 1 (ULK1) or UV radiation resistance-associated gene protein (UVRAG), among others [73][74][75][76][77]. Inversely, in nutrient-poor conditions, reduced mTORC1 activity induces autophagy, which leads to the removal of proteins and organelles to compensate for nutrient starvation. As it described previously, a reduction in glucose levels induces AMPK activation, which phosphorylates Raptor, which in turn inhibits mTORC1 and triggers autophagy [72][78][79]. Inactivation of mTORC1 under starvation conditions prevents the maintenance of Ser757 phosphorylation of ULK1, which induces autophagy initiation [80]. As a general idea, autophagy induction after ischemia could be considered an intrinsic mechanism of neuroprotection. In this regard, some experimental evidence confirms this hypothesis [78]. The pharmacological induction of autophagy after ischemia using rapamycin reduces brain damage [72][81][82][83]. However, other results indicate that excessive autophagic flow aggravates ischemic damage [84]. Therefore, autophagy is another example of a cellular mechanism that requires fine regulation to have positive or negative effects after injury.
The mTOR activity plays a pivotal role in axonal growth, a highly regulated process involved in synaptic plasticity and development [85]. Rapamycin administration to primary cultures of neurons induces the inhibition of axonal growth and prevents neuronal differentiation [30]. However, mTORC1 activation by ablation of their negative upstream regulators (TSC or PTEN) stimulates regenerative processes related with axon guidance and growth [86][87]. In this regard, the presence of some components of the mTORC1 pathway, including P70S6K and 4EBP-1, has been reported in the axonal cones of primary neurons. This interesting observation reflects the importance of mTORC1 location in the appropriate subcellular compartment to regulate certain neuron-specific mechanisms, such as axonal growth, in situ [26].
The downstream targets of mTORC2 include several members of the AGC kinase family, such as Akt, PKC, and SGK1 [88] (Figure 4). As mentioned previously, mTORC2 participates in neuronal cytoskeleton remodeling, specifically in the actin cytoskeleton [8]. Rictor ablation induces a reduction in mTORC2 activity, thereby having impacts on neuronal size and morphology [89][90]. Indeed, cytoskeleton rearrangement is a crucial factor for synaptic plasticity [91].
References
- Van Dam, T.J.; Zwartkruis, F.J.; Bos, J.L.; Snel, B. Evolution of the TOR pathway. J. Mol. Evol. 2011, 73, 209–220.
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976.
- Bercury, K.K.; Dai, J.; Sachs, H.H.; Ahrendsen, J.T.; Wood, T.L.; Macklin, W.B. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J. Neurosci. 2014, 34, 4466–4480.
- Picard, K.; St-Pierre, M.-K.; Vecchiarelli, H.A.; Bordeleau, M.; Tremblay, M.-È. Neuroendocrine, neuroinflammatory and pathological outcomes of chronic stress: A story of microglial remodeling. Neurochem. Int. 2021, 145, 104987.
- Karalis, V.; Bateup, H.S. Current Approaches and Future Directions for the Treatment of mTORopathies. Dev. Neurosci. 2021, 43, 143–158.
- Moloney, P.B.; Cavalleri, G.L.; Delanty, N. Epilepsy in the mTORopathies: Opportunities for precision medicine. Brain Commun. 2021, 3, fcab222.
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223.
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128.
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168.
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743.
- Gong, L.; Shu, J.; Chen, X.; Pan, H.; Chen, G.; Bi, Y.; Cui, D.; Li, X.; Liu, D.; Wang, L.; et al. DEPTOR inhibits lung tumorigenesis by inactivating the EGFR-mTOR signals. Cancer Lett. 2021, 519, 263–276.
- Gagné, L.M.; Morin, N.; Lavoie, N.; Bisson, N.; Lambert, J.P.; Mallette, F.A.; Huot, M. Tyrosine phosphorylation of DEPTOR functions as a molecular switch to activate mTOR signaling. J. Biol. Chem. 2021, 297, 101291.
- Wälchli, M.; Berneiser, K.; Mangia, F.; Imseng, S.; Craigie, L.M.; Stuttfeld, E.; Hall, M.N.; Maier, T. Regulation of human mTOR complexes by DEPTOR. Elife 2021, 10, e70781.
- Wang, T.; Blumhagen, R.; Lao, U.; Kuo, Y.; Edgar, B.A. LST8 regulates cell growth via target-of-rapamycin complex 2 (TORC2). Mol. Cell Biol. 2012, 32, 2203–2213.
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871.
- Ghomlaghi, M.; Yang, G.; Shin, S.Y.; James, D.E.; Nguyen, L.K. Dynamic modelling of the PI3K/MTOR signalling network uncovers biphasic dependence of mTORC1 activity on the mTORC2 subunit SIN1. PLoS Comput. Biol. 2021, 17, e1008513.
- Sini, P.; James, D.; Chresta, C.; Guichard, S. Simultaneous inhibition of mTORC1 and mTORC2 by mTOR kinase inhibitor AZD8055 induces autophagy and cell death in cancer cells. Autophagy 2010, 6, 553–554.
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203.
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870.
- Perez-Alvarez, M.J.; Villa Gonzalez, M.; Benito-Cuesta, I.; Wandosell, F.G. Role of mTORC1 Controlling Proteostasis after Brain Ischemia. Front. Neurosci. 2018, 12, 60.
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293.
- Shiota, C.; Woo, J.T.; Lindner, J.; Shelton, K.D.; Magnuson, M.A. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev. Cell 2006, 11, 583–589.
- Gangloff, Y.G.; Mueller, M.; Dann, S.G.; Svoboda, P.; Sticker, M.; Spetz, J.F.; Um, S.H.; Brown, E.J.; Cereghini, S.; Thomas, G.; et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell Biol. 2004, 24, 9508–9516.
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137.
- Murakami, M.; Ichisaka, T.; Maeda, M.; Oshiro, N.; Hara, K.; Edenhofer, F.; Kiyama, H.; Yonezawa, K.; Yamanaka, S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell Biol. 2004, 24, 6710–6718.
- Lipton, J.O.; Sahin, M. The neurology of mTOR. Neuron 2014, 84, 275–291.
- Bockaert, J.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187.
- Lee, D.Y. Roles of mTOR Signaling in Brain Development. Exp. Neurobiol. 2015, 24, 177–185.
- Carroll, B. Spatial regulation of mTORC1 signalling: Beyond the Rag GTPases. Semin. Cell Dev. Biol. 2020, 107, 103–111.
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular neurobiology of mTOR. Neuroscience 2017, 341, 112–153.
- Gao, Y.; Moten, A.; Lin, H.-K. Akt: A new activation mechanism. Cell Res. 2014, 24, 785–786.
- Thiebaut, A.M.; Buendia, I.; Ginet, V.; Lemarchand, E.; Boudjadja, M.B.; Hommet, Y.; Lebouvier, L.; Lechevallier, C.; Maillasson, M.; Hedou, E.; et al. Thrombolysis by PLAT/tPA increases serum free IGF1 leading to a decrease of deleterious autophagy following brain ischemia. Autophagy 2021, 1–21.
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590.
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713.
- Muraleedharan, R.; Dasgupta, B. AMPK in the brain: Its roles in glucose and neural metabolism. FEBS J. 2021.
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575.
- Momcilovic, M.; Hong, S.P.; Carlson, M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J. Biol. Chem. 2006, 281, 25336–25343.
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19.
- Xu, J.; Ji, J.; Yan, X.H. Cross-talk between AMPK and mTOR in regulating energy balance. Crit. Rev. Food Sci. Nutr. 2012, 52, 373–381.
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812.
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226.
- Laplante, M.; Sabatini, D.M. mTOR Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011593.
- Seong, M.; Lee, J.; Kang, H. Hypoxia-induced regulation of mTOR signaling by miR-7 targeting REDD1. J. Cell Biochem. 2019, 120, 4523–4532.
- Shoshani, T.; Faerman, A.; Mett, I.; Zelin, E.; Tenne, T.; Gorodin, S.; Moshel, Y.; Elbaz, S.; Budanov, A.; Chajut, A.; et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell Biol. 2002, 22, 2283–2293.
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251.
- Tan, C.Y.; Hagen, T. mTORC1 dependent regulation of REDD1 protein stability. PLoS ONE 2013, 8, e63970.
- Lee, C.H.; Park, J.H.; Cho, J.H.; Ahn, J.H.; Yan, B.C.; Lee, J.C.; Shin, M.C.; Cheon, S.H.; Cho, Y.S.; Cho, J.H.; et al. Changes and expressions of Redd1 in neurons and glial cells in the gerbil hippocampus proper following transient global cerebral ischemia. J. Neurol. Sci. 2014, 344, 43–50.
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945.
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139.
- Narita, M.; Inoki, K. Rags connect mTOR and autophagy. Small GTPases 2012, 3, 111–114.
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501.
- Rabanal-Ruiz, Y.; Korolchuk, V.I. mTORC1 and Nutrient Homeostasis: The Central Role of the Lysosome. Int. J. Mol. Sci. 2018, 19, 818.
- Kimball, S.R.; Jefferson, L.S. Molecular mechanisms through which amino acids mediate signaling through the mammalian target of rapamycin. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 39–44.
- Takei, N.; Furukawa, K.; Hanyu, O.; Sone, H.; Nawa, H. A possible link between BDNF and mTOR in control of food intake. Front. Psychol. 2014, 5, 1093.
- Ishizuka, Y.; Kakiya, N.; Witters, L.A.; Oshiro, N.; Shirao, T.; Nawa, H.; Takei, N. AMP-activated protein kinase counteracts brain-derived neurotrophic factor-induced mammalian target of rapamycin complex 1 signaling in neurons. J. Neurochem. 2013, 127, 66–77.
- Boutouja, F.; Stiehm, C.M.; Platta, H.W. mTOR: A Cellular Regulator Interface in Health and Disease. Cells 2019, 8, 18.
- Böhm, R.; Imseng, S.; Jakob, R.P.; Hall, M.N.; Maier, T.; Hiller, S. The dynamic mechanism of 4E-BP1 recognition and phosphorylation by mTORC1. Mol. Cell 2021, 81, 2403–2416.e5.
- Batool, A.; Majeed, S.T.; Aashaq, S.; Majeed, R.; Shah, G.; Nazir, N.; Andrabi, K.I. Eukaryotic Initiation Factor 4E (eIF4E) sequestration mediates 4E-BP1 response to rapamycin. Int. J. Biol. Macromol. 2019, 125, 651–659.
- Ayuso, M.I.; Hernández-Jiménez, M.; Martín, M.E.; Salinas, M.; Alcázar, A. New hierarchical phosphorylation pathway of the translational repressor eIF4E-binding protein 1 (4E-BP1) in ischemia-reperfusion stress. J. Biol. Chem. 2010, 285, 34355–34363.
- Ayuso, M.I.; Martinez-Alonso, E.; Salvador, N.; Bonova, P.; Regidor, I.; Alcázar, A. Dissociation of eIF4E-binding protein 2 (4E-BP2) from eIF4E independent of Thr37/Thr46 phosphorylation in the ischemic stress response. PLoS ONE 2015, 10, e0121958.
- Gingras, A.C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001, 15, 2852–2864.
- Laplante, M.; Sabatini, D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol. 2009, 19, R1046–R1052.
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28.
- Katsu-Jiménez, Y.; Alves, R.M.P.; Giménez-Cassina, A. Food for thought: Impact of metabolism on neuronal excitability. Exp. Cell Res. 2017, 360, 41–46.
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183.
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236.
- Yamauchi, Y.; Furukawa, K.; Hamamura, K.; Furukawa, K. Positive feedback loop between PI3K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: Induction of the lipogenic pathway by a melanoma antigen. Cancer Res. 2011, 71, 4989–4997.
- Lamming, D.W.; Sabatini, D.M. A Central role for mTOR in lipid homeostasis. Cell Metab. 2013, 18, 465–469.
- Schüle, M.; Butto, T.; Dewi, S.; Schlichtholz, L.; Strand, S.; Gerber, S.; Endres, K.; Schweiger, S.; Winter, J. mTOR Driven Gene Transcription Is Required for Cholesterol Production in Neurons of the Developing Cerebral Cortex. Int. J. Mol. Sci. 2021, 22, 6034.
- Norrmén, C.; Figlia, G.; Lebrun-Julien, F.; Pereira, J.A.; Trötzmüller, M.; Köfeler, H.C.; Rantanen, V.; Wessig, C.; van Deijk, A.L.; Smit, A.B.; et al. mTORC1 controls PNS myelination along the mTORC1-RXRγ-SREBP-lipid biosynthesis axis in Schwann cells. Cell Rep. 2014, 9, 646–660.
- Thiebaut, A.M.; Hedou, E.; Marciniak, S.J.; Vivien, D.; Roussel, B.D. Proteostasis During Cerebral Ischemia. Front. Neurosci. 2019, 13, 637.
- Ahsan, A.; Liu, M.; Zheng, Y.; Yan, W.; Pan, L.; Li, Y.; Ma, S.; Zhang, X.; Cao, M.; Wu, Z.; et al. Natural compounds modulate the autophagy with potential implication of stroke. Acta Pharm. Sin. B 2021, 11, 1708–1720.
- Munson, M.J.; Allen, G.F.; Toth, R.; Campbell, D.G.; Lucocq, J.M.; Ganley, I.G. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015, 34, 2272–2290.
- Xiang, H.; Zhang, J.; Lin, C.; Zhang, L.; Liu, B.; Ouyang, L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm. Sin. B 2020, 10, 569–581.
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991.
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162.
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216.
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100.
- Shi, Q.; Cheng, Q.; Chen, C. The Role of Autophagy in the Pathogenesis of Ischemic Stroke. Curr. Neuropharmacol. 2021, 19, 629–640.
- Wang, Y.; Zhang, H. Regulation of Autophagy by mTOR Signaling Pathway. Adv. Exp. Med. Biol. 2019, 1206, 67–83.
- Urbanek, T.; Kuczmik, W.; Basta-Kaim, A.; Gabryel, B. Rapamycin induces of protective autophagy in vascular endothelial cells exposed to oxygen-glucose deprivation. Brain Res. 2014, 1553, 1–11.
- Wu, M.; Zhang, H.; Kai, J.; Zhu, F.; Dong, J.; Xu, Z.; Wong, M.; Zeng, L.H. Rapamycin prevents cerebral stroke by modulating apoptosis and autophagy in penumbra in rats. Ann. Clin. Transl. Neurol. 2018, 5, 138–146.
- Zhao, Y.; Zhang, X.; Chen, X.; Wei, Y. Neuronal injuries in cerebral infarction and ischemic stroke: From mechanisms to treatment. Int. J. Mol. Med. 2022, 49, 5070.
- Liu, T.; Han, S.; Dai, Q.; Zheng, J.; Liu, C.; Li, S.; Li, J. IL-17A-Mediated Excessive Autophagy Aggravated Neuronal Ischemic Injuries via Src-PP2B-mTOR Pathway. Front. Immunol. 2019, 10, 2952.
- Jaworski, J.; Sheng, M. The growing role of mTOR in neuronal development and plasticity. Mol. Neurobiol. 2006, 34, 205–219.
- Nie, D.; Di Nardo, A.; Han, J.M.; Baharanyi, H.; Kramvis, I.; Huynh, T.; Dabora, S.; Codeluppi, S.; Pandolfi, P.P.; Pasquale, E.B.; et al. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat. Neurosci. 2010, 13, 163–172.
- Lu, Y.; Belin, S.; He, Z. Signaling regulations of neuronal regenerative ability. Curr. Opin. Neurobiol. 2014, 27, 135–142.
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159.
- Thomanetz, V.; Angliker, N.; Cloëtta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Rüegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308.
- Angliker, N.; Rüegg, M.A. In vivo evidence for mTORC2-mediated actin cytoskeleton rearrangement in neurons. Bioarchitecture 2013, 3, 113–118.
- Huang, W.; Zhu, P.J.; Zhang, S.; Zhou, H.; Stoica, L.; Galiano, M.; Krnjević, K.; Roman, G.; Costa-Mattioli, M. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat. Neurosci. 2013, 16, 441–448.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
05 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No