Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cheol-Hee Kim | -- | 1911 | 2022-04-08 07:09:41 | | | |
| 2 | Rita Xu | Meta information modification | 1911 | 2022-04-08 08:12:06 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kim, C.; Son, M.; Kim, D. Rare Neurological Disorders in Zebrafish. Encyclopedia. Available online: https://encyclopedia.pub/entry/21492 (accessed on 22 June 2026).
Kim C, Son M, Kim D. Rare Neurological Disorders in Zebrafish. Encyclopedia. Available at: https://encyclopedia.pub/entry/21492. Accessed June 22, 2026.
Kim, Cheol-Hee, Myeongjoo Son, Daeyu Kim. "Rare Neurological Disorders in Zebrafish" Encyclopedia, https://encyclopedia.pub/entry/21492 (accessed June 22, 2026).
Kim, C., Son, M., & Kim, D. (2022, April 08). Rare Neurological Disorders in Zebrafish. In Encyclopedia. https://encyclopedia.pub/entry/21492
Kim, Cheol-Hee, et al. "Rare Neurological Disorders in Zebrafish." Encyclopedia. Web. 08 April, 2022.
Copy Citation
Rare diseases are those which affect a small number of people compared to the general population. However, many patients with a rare disease remain undiagnosed, and a large majority of rare diseases still have no form of viable treatment. Approximately 40% of rare diseases include neurologic and neurodevelopmental disorders. In order to understand the characteristics of rare neurological disorders and identify causative genes, various model organisms have been utilized extensively.
rare diseases
neurological rare diseases
undiagnosed diseases
1. Introduction
Rare diseases affect a relatively small portion of people compared to other prevalent diseases; thus, characteristic issues arise, due to their rarity [1]. Although the definition of a rare disease varies slightly from country to country, the United States Congress defined a rare disease in the Orphan Drug Act of 1983 as a condition affecting fewer than 200,000 patients. The total number of Americans suffering from rare diseases is estimated at 25–30 million, while 30 million people are affected by rare disease across Europe [1].
Globally, there are about 8000 rare diseases, including genetic disorders, rare cancers, auto-immune disease, and infectious diseases, which are often serious, progressive, and chronic conditions [2]. Many rare diseases show a variety of signs starting at birth or childhood, including proximal spinal muscular atrophy, neurofibromatosis, chondrodysplasia osteogenesis imperfecta, and Rett syndrome. Rare diseases which appear during adulthood include those such as Huntington diseases, Crohn diseases, amyotrophic lateral sclerosis, and Charcot–Marie–Tooth diseases.
The exact cause of many rare diseases is still unknown, but a large majority of them (~80%) have a genetic cause, including a direct single gene, multifactorial, or chromosome changes [3]. In some cases, genetic changes causing disease are passed from one generation to the next. In other cases, they randomly occur in a person who is the first in a family to receive diagnosis [3]. Many patients with rare conditions undergo a “diagnostic odyssey”, trying various clinical approaches and comprehensive biochemical and genetic tests in hopes of an accurate assessment; however, often, they must wait years for a definitive diagnosis. National and international researchers have made progress in learning how to diagnose, clinically treat, and even prevent many rare diseases (Table 1). Unfortunately, 30% of child patients, which make up 50% of rare-disease patients, die before the age of 5 [3], while approximately 40% of known rare diseases include neurologic and neurodevelopmental disorders.
Table 1. List of rare disease research program or network.
| Program/Network Name | Goals | Homepage Address |
|---|---|---|
| Genetic and Rare Diseases Information Center |
Providing the general public with the latest information on various rare diseases in an easy-to-understand manner. | https://rarediseases.info.nih.gov/diseases |
| International rare disease research consortium |
Contributing to the development of new treatments for rare diseases and methods to uncover the genetic causes of rare diseases. | https://irdirc.org/ |
| National Organization for Rare Disorders |
Raising awareness of rare diseases and improve access to treatment and medical services for patients and their families. | https://rarediseases.org |
| Orphanet (global network) |
Providing international reference knowledge base for rare diseases and orphan drugs. | https://www.orpha.net/ |
| Providing the scientific datasets (research, clinical trials, drugs, etc.) related to rare diseases and orphan drugs. | http://www.orphadata.org/ | |
| Rare Diseases Clinical Research Network |
Providing support for clinical studies and facilitating collaboration, study enrollment, and data sharing. | https://ncats.nih.gov/rdcrn |
| Undiagnosed Diseases Network |
Accelerating identification of genetic causes of rare diseases by validating candidate genes, using model organisms. | https://undiagnosed.hms.harvard.edu/research |
2. Model Organisms for Rare Disease Research
In vitro approaches that use mainly cell or tissue culture can help predict clinical outcomes [4] but are limited in mimicking rare human diseases. The selection of an appropriate model organism is critical for preclinical research. Several important factors of consideration include species similarity to humans (i.e., the closer the phylogeny, the more similar the genetic composition, anatomy, and physiology), genetic homogeneity, a priori knowledge, cost, availability, translatability of results, ease of operation, ethical implications, etc. [5].
Over the past few years, clustered regularly interspaced repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) system (CRISPR/Cas9) genome editing technology has transformed the field and has greatly expanded the repertoire of cell/animal systems available for rare disease modeling [6]. The use of this genome editing technology with genetic model organisms has enhanced the understanding of human rare diseases. For example, mouse and zebrafish models, at first glance, appear completely unrelated to humans; however, on the genetic and physiological level, they respectively share about 85% and 71% of the same genes and possess major organ systems in common, such as the central nervous system, circulatory system, digestive system, etc. To study the functional consequences of the hundreds of rare variants discovered by genome sequencing, researchers have developed the use of specific model organisms, including roundworms (C. elegans), fruit flies (Drosophila melanogaster), and zebrafish (Danio rerio).
2.1. Caenorhabditis elegans (C. elegans)
The significance of non-mammalian model organisms has been recognized for quite some time [1][6]. The Nobel Prize in Physiology or Medicine has been awarded to researchers for their discoveries in apoptosis (2022) and RNA interference using worms (2006).
C. elegans is an unsegmented pseudocoelomate, lacking circulatory and respiratory systems, whose body segments are serially repeated one after the other [7]. C. elegans was used primarily for neuronal development research in 1963 and has since seen extensive use as a model organism for research into neural and molecular mechanisms of learning, memory, mating behavior, chemotaxis, thermotaxis, and mechano-transduction [8]. In addition, the C. elegans model has provided insights into finer mechanistic details of human health, including inter-cellular signaling pathways (e.g., Notch signaling), intra-cellular pathways (e.g., autophagy), molecular machines (e.g., spliceosome), and multi-cellular processes (e.g., basement membrane biology) [9].
In 2019, the first C. elegans multicellular organism underwent whole-genome sequencing and is the only organism to have completed its connectome (the “wiring diagram” of neurons) [10][11][12]. There are 20,512 protein-coding genes [13], and 83% of the worm proteome is found to have human homologous genes [14]. Only 11% or less contain roundworm-specific genes. These findings provide the basis by which C. elegans has served as a suitable model organism for human gene functional research [15][16][17][18]. In the case of rare disease modeling, C. elegans provides an ideal system to study human diseases.
2.2. Fruit Fly (Drosophila melanogaster)
Drosophila melanogaster is a species of fly in the Drosophila family (taxonomic order Diptera). For the past century, the Drosophila melanogaster has been used as a model organism to understand the fundamentals of genetics, developmental biology, immunity, and neuroscience [19][20]. Five Nobel Prizes have been awarded to fruit fly scientists for their work with the animal in 2017. Drosophila melanogaster has emerged as an important model system for dissecting and understanding the molecular mechanisms underlying rare human diseases, due to its rapid life cycle, relatively simple genetics (only four pairs of chromosomes), and large number of offspring per generation. This rise is owed in part to the first genome-wide survey of ~1000 genes registered in the Online Mendelian Inheritance in Man (OMIM), which found that 75% of disease-causing genes in humans are conserved in Drosophila [21]. Of the approximately 4000 human disease-related genes currently displayed in OMIM, ~85% have homologues in Drosophila. Considering that ~65% of protein-coding genes are conserved between humans and flies, the data suggest that genes conserved between these species are more likely to implicate genetic disease in humans [22][23]. In addition to being used as a tool for dissecting rare disease mechanisms and exploring potential therapeutic avenues, flies have emerged as a key tool in explaining variants of uncertain significance found in patients [24][25].
2.3. Zebrafish (Danio rerio)
The zebrafish (Danio rerio) is a small freshwater fish, 3-to-4 cm long, with a lifespan of about 2 years. Zebrafish have become increasingly important in scientific research since the 1960s, due to several distinct advantages over other vertebrate models.
Because of the simplicity of their natural habitat, lab maintenance of a zebrafish colony is much easier than simulating the conditions necessary for mammals. Therefore, zebrafish can be grown in a cost-effective manner. Their short generation time of 3 months helps accelerate experimental progress [26], and ex utero development facilitates the observation and rapid experimental manipulation of embryos. In addition, zebrafish have large clutch sizes, ranging from 200 to 300 embryos per adult mating pair, which ensures a robust stock of animals for research work. Due to these features, combined with the relatively small size of the embryo/larva/adult, zebrafish are well-suited for high-throughput screening of potential neuroactive compounds.
The zebrafish possesses many characteristics that make it an invaluable model to study human diseases [27]; however, one of its unique advantages is the unparalleled optical clarity of the embryo, which allows for the visualization of individual genes (fluorescently stained or labeled) throughout development, using non-invasive imaging techniques [28][29][30][31] (Figure 1A). This transparency of the embryo also facilitates genetic manipulation, such that gene function can readily be studied by the injection of synthetic mRNA or plasmid DNA into early stage zebrafish embryos generating transgenic zebrafish lines or altering gene function through genome editing techniques, such as the inclusion of zinc finger nucleases (ZFNs), transcription activation-like effector nucleases (TALENs), and the clustered regularly interspaced repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) system [32][33][34]. Furthermore, the zebrafish genome has been sequenced, and 71% of human genes and 82% of human disease-related genes have orthologs in zebrafish [35]. Both ZFNs and TALENs require the creation of customized protein compositions for each target site, and these systems are not suitable for large-scale applications. However, the CRISPR/Cas9 system relies on target-site recognition by a custom guide RNA (gRNA) molecule and requires only one oligonucleotide to be designed for each target site. The success of CRISPR-mediated transgenic transformation in zebrafish can largely be attributed to frame-shift-generating null alleles via non-homologous extremity joint (NHEJ)-mediated CRISPR-induced DNA break repair [35].
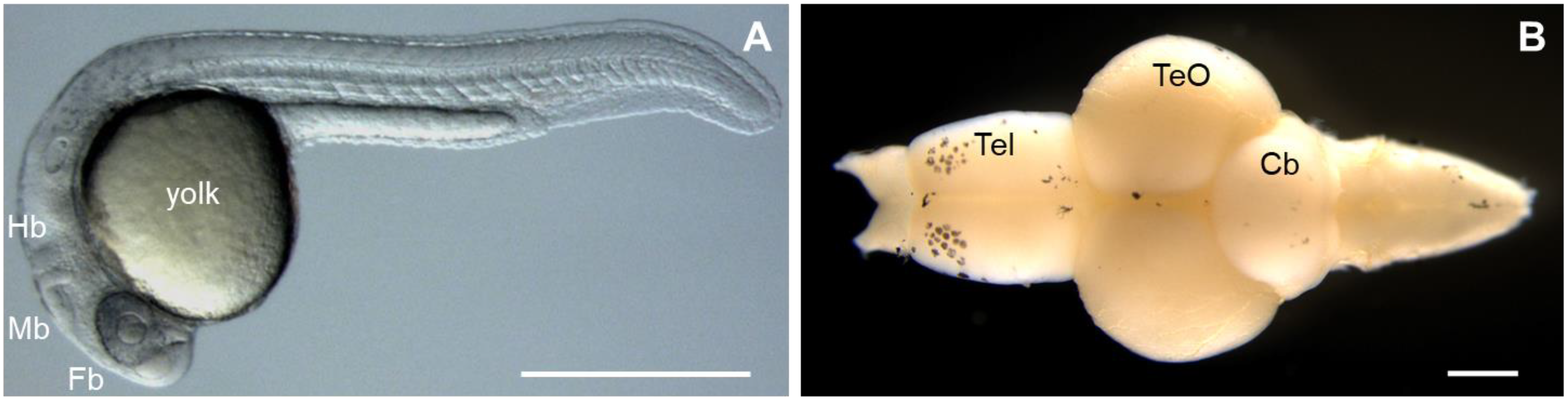
Figure 1. Major features of the zebrafish (Danio rerio). (A) Zebrafish embryo at day 1 of development. Fb, forebrain; Mb, midbrain; Hb, hindbrain. (B) Dissection of brain from an adult zebrafish. Tel, telencephalon; TeO, optic tectum; Cb, cerebellum. Rostral part of the brain is pointing to the left. Scale bars: 500 µm.
Interestingly, similarities between the zebrafish and human nervous system (anatomy and physiological signaling) have been reported [33]. The zebrafish brain is composed of the forebrain (telencephalon), midbrain (optic tectum), and hindbrain (cerebellum) (Figure 1B); and many cells, including astrocytes, oligodendrocytes, microglia, cerebellar Purkinje cells, myelin, and motor neurons, are also similar to human cells. Further studies of spinal nerve patterning, neural differentiation, and vertebrate network development in adult zebrafish revealed similarities to higher vertebrates [33]. Due to these characteristics, zebrafish are widely used to validate candidate disease genes and to elucidate the molecular mechanisms and pathophysiology of neurological disease. The continued increase in the use of zebrafish in biomedical research publications reflects their expanding popularity [33] (Table 2).
Table 2. Zebrafish modeling for rare neurological diseases.
| Gene Name | Related Disease | Zebrafish Phenotype | Publication |
|---|---|---|---|
| sod1 | Amyotrophic Lateral Sclerosis | Motor neuron loss Muscle atrophy |
[36] |
| fus | Shortened motor neuron length Decreased neuromuscular junction Impaired motor behavior Decreased life span Increase of the smallest tau transcripts |
[37] | |
| tardbp | Axonopathy of the motor neurons Premature of axonal branch |
[38] | |
| c9orf72 | Impaired motor behavior Cognitive impairment Muscle atrophy Motor neuron loss |
[39] | |
| fam50a | Armfield XLID syndrome | Abnormal neurogenesis Abnormal craniofacial patterning |
[40] |
| dyrk1a | Down Syndrome and Autism | Decreased brain size Increased anxiolytic behavior Impaired social interaction/cohesion |
[41] |
| wdr11 | Idiopathic Hypogonadotropic Hypogonadism Kallmann Syndrome |
Delayed puberty Impaired sense of smell |
[42] |
| eftud2 | Mandibulofacial Dysostosis, Guion–Almeida Type | Decreased brain size Small eyes Curved body Early embryonic lethality |
[43] |
| zc4h2 | Miles–Carpenter Syndrome | Abnormal swimming Increased twitching Motor hyperactivity Eye movement deficits Pectoral fin contractures |
[44] |
| phf21a | Potocki–Shaffer Syndrome | Abnormal head and jaw size Change of head and face shape |
[45] |
| eif4a3 | Richieri–Costa–Pereira Syndrome | Underdevelopment of craniofacial cartilage and bone structures | [46] |
| eif2b5 | Vanishing White Matter Disease | Early embryonic lethality Loss of oligodendrocyte precursor cells Impaired motor behavior |
[47] |
| eif2b3 | Defected myelin gene expression Defected glial cell differentiation |
[48] | |
| sam2 | 12q14.1 Deletion Syndrome | Increased of fear, anxiety-related behaviors, and autism | [49] |
References
- Wangler, M.F.; Yamamoto, S.; Chao, H.T.; Posey, J.E.; Westerfield, M.; Postlethwait, J.; Members of the Undiagnosed Diseases Network (UDN); Hieter, P.; Boycott, K.M.; Campeau, P.M.; et al. Model Organisms Facilitate Rare Disease Diagnosis and Therapeutic Research. Genetics 2017, 207, 9–27.
- The Lancet Neurology. Rare neurological diseases: A united approach is needed. Lancet Neurol. 2011, 10, 109.
- Field, M.J.; Boat, T.F. Rare Diseases and Orphan Products: Accelerating Research and Development; National Academies Press: Washington, DC, USA, 2010; pp. 51–69.
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell. Biol. 2020, 21, 571–584.
- National Research Council (US) Committee on New and Emerging Models in Biomedical and Behavioral Research. Biomedical Models and Resources: Current Needs and Future Opportunities; National Academies Press: Washington, DC, USA, 1998; pp. 24–39.
- Bellen, H.J.; Tong, C.; Tsuda, H. 100 years of Drosophila research and its impact on vertebrate neuroscience: A history lesson for the future. Nat. Rev. Neurosci. 2010, 11, 514–522.
- Wallace, R.L.; Ricci, C.; Melone, G. A cladistic analysis of pseudocoelomate (aschelminth) morphology. Invertebr. Biol. 1996, 115, 104–112.
- Schafer, W.R. Deciphering the neural and molecular mechanisms of C. elegans behavior. Curr. Biol. 2005, 15, R723–R729.
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science Taylor and Francis Group: New York, NY, USA, 2015.
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845.
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291.
- Boycott, K.M.; Rath, A.; Chong, J.X.; Hartley, T.; Alkuraya, F.S.; Baynam, G.; Brookes, A.J.; Brudno, M.; Carracedo, A.; den Dunnen, J.T.; et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 2017, 100, 695–705.
- Girard, L.R.; Fiedler, T.J.; Harris, T.W.; Carvalho, F.; Antoshechkin, I.; Han, M.; Sternberg, P.W.; Stein, L.D.; Chalfie, M. WormBook: The online review of Caenorhabditis elegans biology. Nucleic Acids Res. 2007, 35, D472–D475.
- Lai, C.H.; Chou, C.Y.; Ch’ang, L.Y.; Liu, C.S.; Lin, W. Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 2000, 10, 703–713.
- Kenyon, C. A conserved regulatory system for aging. Cell 2001, 105, 165–168.
- O’Kane, C.J. Modelling human diseases in Drosophila and Caenorhabditis. Semin. Cell Dev. Biol. 2003, 14, 3–10.
- Schulenburg, H.; Kurz, C.L.; Ewbank, J.J. Evolution of the innate immune system: The worm perspective. Immunol. Rev. 2004, 198, 36–58.
- Riessland, M.; Kaczmarek, A.; Schneider, S.; Swoboda, K.J.; Löhr, H.; Bradler, C.; Grysko, V.; Dimitriadi, M.; Hosseinibarkooie, S.; Torres-Benito, L.; et al. Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am. J. Hum. Genet. 2017, 100, 297–315.
- Bellen, H.J.; Yamamoto, S. Morgan’s legacy: Fruit flies and the functional annotation of conserved genes. Cell 2015, 163, 12–14.
- Wangler, M.F.; Yamamoto, S.; Bellen, H.J. Fruit flies in biomedical research. Genetics 2015, 199, 639–653.
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125.
- Wang, J.; Al-Ouran, R.; Hu, Y.; Kim, S.Y.; Wan, Y.W.; Wangler, M.F.; Yamamoto, S.; Chao, H.T.; Comjean, A.; Mohr, S.E.; et al. MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am. J. Hum. Genet. 2017, 100, 843–853.
- Hu, Y.; Flockhart, I.; Vinayagam, A.; Bergwitz, C.; Berger, B.; Perrimon, N.; Mohr, S.E. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinform. 2011, 12, 357.
- Bellen, H.J.; Wangler, M.F.; Yamamoto, S. The fruit fly at the interface of diagnosis and pathogenic mechanisms of rare and common human diseases. Hum. Mol. Genet. 2019, 28, R207–R214.
- Link, N.; Chung, H.; Jolly, A.; Withers, M.; Tepe, B.; Arenkiel, B.R.; Shah, P.S.; Krogan, N.J.; Aydin, H.; Geckinli, B.B.; et al. Mutations in ANKLE2, a ZIKA Virus Target, Disrupt an Asymmetric Cell Division Pathway in Drosophila Neuroblasts to Cause Microcephaly. Dev. Cell. 2019, 51, 713–729.
- Detrich, H.W., III; Westerfield, M.; Zon, L.I. Overview of the zebrafish system. Methods Cell Biol. 1998, 59, 3–10.
- Eisen, J.S. History of zebrafish research. In The Zebrafish in Biomedical Research; Academic Press: Cambridge, MA, USA, 2020; pp. 3–14.
- Kimmel, C.B. Genetics and early development of zebrafish. Trends Genet. 1989, 5, 283–288.
- Solnica-Krezel, L.; Stemple, D.L.; Driever, W. Transparent things: Cell fates and cell movements during early embryogenesis of zebrafish. BioEssays 1995, 17, 931–939.
- Cooper, M.S.; D’Amico, L.A.; Henry, C.A. Analyzing morphogenetic cell behaviors in vitally stained zebrafish embryos. Methods Mol. Biol. 1999, 122, 185–204.
- Spitsbergen, J.M.; Kent, M.L. The state of the art of the zebrafish model for toxicology and toxicologic pathology research—Advantages and current limitations. Toxicol. Pathol. 2003, 31, 62–87.
- Carney, T.J.; Mosimann, C. Switch and Trace: Recombinase Genetics in Zebrafish. Trends Genet. 2018, 34, 362–378.
- Adamson, K.I.; Sheridan, E.; Grierson, A.J. Use of zebrafish models to investigate rare human disease. J. Med. Genet. 2018, 55, 641–649.
- Ahmad, G.; Amiji, M. Use of CRISPR/Cas9 gene-editing tools for developing models in drug discovery. Drug Discov. Today 2018, 23, 519–533.
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503.
- Ramesh, T.; Lyon, A.N.; Pineda, R.H.; Wang, C.; Janssen, P.M.; Canan, B.D.; Burghes, A.H.; Beattie, C.E. A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis. Models Mech. 2010, 3, 652–662.
- Bourefis, A.R.; Campanari, M.L.; Buee-Scherrer, V.; Kabashi, E. Functional characterization of a FUS mutant zebrafish line as a novel genetic model for ALS. Neurobiol. Dis. 2020, 142, 104935.
- Hewamadduma, C.A.; Grierson, A.J.; Ma, T.P.; Pan, L.; Moens, C.B.; Ingham, P.W.; Ramesh, T.; Shaw, P.J. Tardbpl splicing rescues motor neuron and axonal development in a mutant tardbp zebrafish. Hum. Mol. Genet. 2013, 22, 2376–2386.
- Shaw, M.P.; Higginbottom, A.; McGown, A.; Castelli, L.M.; James, E.; Hautbergue, G.M.; Shaw, P.J.; Ramesh, T.M. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 2018, 6, 125.
- Lee, Y.R.; Khan, K.; Armfield-Uhas, K.; Srikanth, S.; Thompson, N.A.; Pardo, M.; Yu, L.; Norris, J.W.; Peng, Y.; Gripp, K.W.; et al. Mutations in FAM50A suggest that Armfield XLID syndrome is a spliceosomopathy. Nat. Commun. 2020, 11, 3698.
- Kim, O.H.; Cho, H.J.; Han, E.; Hong, T.I.; Ariyasiri, K.; Choi, J.H.; Hwang, K.S.; Jeong, Y.M.; Yang, S.Y.; Yu, K.; et al. Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism 2017, 8, 50.
- Kim, H.G.; Ahn, J.W.; Kurth, I.; Ullmann, R.; Kim, H.T.; Kulharya, A.; Ha, K.S.; Itokawa, Y.; Meliciani, I.; Wenzel, W.; et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2010, 87, 465–479.
- Deml, B.; Reis, L.M.; Muheisen, S.; Bick, D.; Semina, E.V. EFTUD2 deficiency in vertebrates: Identification of a novel human mutation and generation of a zebrafish model. Birth Defects Res. A Clin. Mol. Teratol. 2015, 103, 630–640.
- May, M.; Hwang, K.S.; Miles, J.; Williams, C.; Niranjan, T.; Kahler, S.G.; Chiurazzi, P.; Steindl, K.; Van Der Spek, P.J.; Swagemakers, S.; et al. ZC4H2, an XLID gene, is required for the generation of a specific subset of CNS interneurons. Hum. Mol. Genet. 2015, 24, 4848–4861.
- Kim, H.G.; Kim, H.T.; Leach, N.T.; Lan, F.; Ullmann, R.; Silahtaroglu, A.; Kurth, I.; Nowka, A.; Seong, I.S.; Shen, Y.; et al. Translocations disrupting PHF21A in the Potocki-Shaffer-syndrome region are associated with intellectual disability and craniofacial anomalies. Am. J. Hum. Genet. 2012, 91, 56–72.
- Favaro, F.P.; Alvizi, L.; Zechi-Ceide, R.M.; Bertola, D.; Felix, T.M.; de Souza, J.; Raskin, S.; Twigg, S.R.; Weiner, A.M.; Armas, P.; et al. A noncoding expansion in EIF4A3 causes Richieri-Costa-Pereira syndrome, a craniofacial disorder associated with limb defects. Am. J. Hum. Genet. 2014, 94, 120–128.
- Keefe, M.D.; Soderholm, H.E.; Shih, H.Y.; Stevenson, T.J.; Glaittli, K.A.; Bowles, D.M.; Scholl, E.; Colby, S.; Merchant, S.; Hsu, E.W.; et al. Vanishing white matter disease expression of truncated EIF2B5 activates induced stress response. eLife. 2020, 9, e56319.
- Lee, Y.R.; Kim, S.H.; Ben-Mahmoud, A.; Kim, O.H.; Choi, T.I.; Lee, K.H.; Ku, B.; Eum, J.; Kee, Y.; Lee, S.; et al. Eif2b3 mutants recapitulate phenotypes of vanishing white matter disease and validate novel disease alleles in zebrafish. Hum. Mol. Genet. 2021, 30, 331–342.
- Choi, J.H.; Jeong, Y.M.; Kim, S.; Lee, B.; Ariyasiri, K.; Kim, H.T.; Jung, S.H.; Hwang, K.S.; Choi, T.I.; Park, C.O.; et al. Targeted knockout of a chemokine-like gene increases anxiety and fear responses. Proc. Natl. Acad. Sci. USA 2018, 115, E1041–E1050.
More
Information
Subjects:
Developmental Biology; Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
908
Revisions:
2 times
(View History)
Update Date:
08 Apr 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No