 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stéphanie Tomé | -- | 3505 | 2022-04-07 00:39:11 | | | |
| 2 | Yvaine Wei | -4 word(s) | 3501 | 2022-04-07 05:26:07 | | | | |
| 3 | Yvaine Wei | Meta information modification | 3501 | 2022-04-07 05:27:07 | | |
Video Upload Options
Among the trinucleotide repeat disorders, myotonic dystrophy type 1 (DM1) is one of the most complex neuromuscular diseases caused by an unstable CTG repeat expansion in the DMPK gene. DM1 patients exhibit high variability in the dynamics of CTG repeat instability and in the manifestations and progression of the disease. The largest expanded alleles are generally associated with the earliest and most severe clinical form. However, CTG repeat length alone is not sufficient to predict disease severity and progression, suggesting the involvement of other factors. Several data support the role of epigenetic alterations in clinical and genetic variability observed in DM1.
1. Introduction

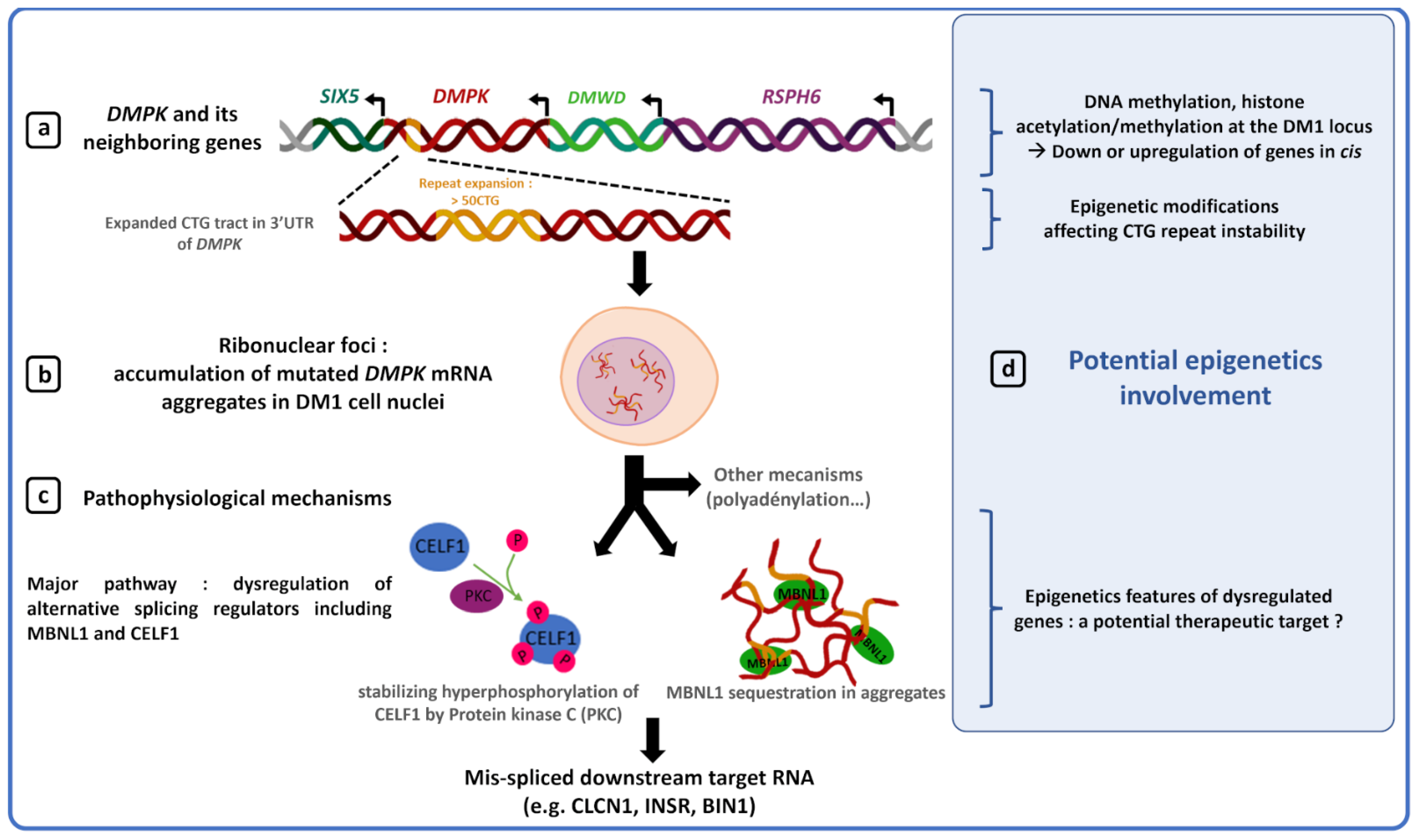
2. Epigenetic Modifications and DM1 Clinical Features
2.1. CDM1 Form Associated with Hypermethylation of the DM1 Locus and Large CTG Repeat Expansions
2.2. DM1 Symptoms Associated with Specific Epigenetic Patterns
3. Sex, CTG Repeats and Epigenetics
3.1. DNA Methylation at the DMPK Locus May Contribute to Sex Differences in DM1 Symptoms
3.2. DNA Methylation Alters the Dynamics of CTG Repeat Instability- Role in the DM1 Phenotype?
3.3. DM1 Locus Methylation Deregulated in Patients with an Interrupted Expanded Allele and Less Severe Symptoms
4. Epigenetics, Gene Expression and Chromatin at the DM1 Locus
5. Epigenetic and Therapy for DM1
5.1. Modulation of Somatic Mosaicism by Global Demethylation in DM1 Models
5.2. Modulation of CTG Somatic Instability by Inhibition of Histone Deacetylase
In the same model, Williams et al. demonstrated that RGFP966, a selective HDAC3 inhibitor, affects repeat instability by deacetylating MSH3 protein, a protein involved in the formation of repeat expansions in DM1 mouse models [40][85][86]. Inhibition of HDACs could contribute to attenuate DM1 disease worsening by modulating repeat instability or another pathway that needs to be identified. However, further studies in murine models are needed to ensure that the use of HDAC inhibitors is truly beneficial to the patient without inducing deleterious side effects.
5.3. Modulation of MBNL1 Transcription by a Methyl Sensitive Enhancer
6. Conclusions
Although promising, the use of methylation as biomarker to refine prognosis and improve treatments must be taken with caution as various external and internal factors such as the environment or the patient’s age at the time of sampling are known to affect methylation levels [11][23][97][98][99][100]. Chromatin structure and gene expression modulated by epigenetic processes are less studied in DM1. It is not yet known whether these parameters could be used as biomarkers in DM1. However, they remain interesting potential therapeutic targets to alleviate DM1 symptoms by epigenetic regulation of activator or repressor of dysfunctional DM1 proteins (MBNL1) or reduction of somatic instability (HDAC inhibitors) [83][84][86][89][91].
References
- Morrison, B.M. Neuromuscular Diseases. Semin. Neurol. 2016, 36, 409–418.
- Khristich, A.N.; Mirkin, S.M. On the Wrong DNA Track: Molecular Mechanisms of Repeat-Mediated Genome Instability. J. Biol. Chem. 2020, 295, 4134–4170.
- Johnson, N.E.; Butterfield, R.J.; Mayne, K.; Newcomb, T.; Imburgia, C.; Dunn, D.; Duval, B.; Feldkamp, M.L.; Weiss, R.B. Population-Based Prevalence of Myotonic Dystrophy Type 1 Using Genetic Analysis of Statewide Blood Screening Program. Neurology 2021, 96, e1045–e1053.
- De Antonio, M.; Dogan, C.; Hamroun, D.; Mati, M.; Zerrouki, S.; Eymard, B.; Katsahian, S.; Bassez, G. French Myotonic Dystrophy Clinical Network Unravelling the Myotonic Dystrophy Type 1 Clinical Spectrum: A Systematic Registry-Based Study with Implications for Disease Classification. Rev. Neurol. 2016, 172, 572–580.
- Aslanidis, C.; Jansen, G.; Amemiya, C.; Shutler, G.; Mahadevan, M.; Tsilfidis, C.; Chen, C.; Alleman, J.; Wormskamp, N.G.; Vooijs, M. Cloning of the Essential Myotonic Dystrophy Region and Mapping of the Putative Defect. Nature 1992, 355, 548–551.
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T. Molecular Basis of Myotonic Dystrophy: Expansion of a Trinucleotide (CTG) Repeat at the 3’ End of a Transcript Encoding a Protein Kinase Family Member. Cell 1992, 68, 799–808.
- Buxton, J.; Shelbourne, P.; Davies, J.; Jones, C.; Van Tongeren, T.; Aslanidis, C.; de Jong, P.; Jansen, G.; Anvret, M.; Riley, B. Detection of an Unstable Fragment of DNA Specific to Individuals with Myotonic Dystrophy. Nature 1992, 355, 547–548.
- Harley, H.G.; Rundle, S.A.; Reardon, W.; Myring, J.; Crow, S.; Brook, J.D.; Harper, P.S.; Shaw, D.J. Unstable DNA Sequence in Myotonic Dystrophy. Lancet 1992, 339, 1125–1128.
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K. Myotonic Dystrophy Mutation: An Unstable CTG Repeat in the 3’ Untranslated Region of the Gene. Science 1992, 255, 1253–1255.
- Lanni, S.; Pearson, C.E. Molecular Genetics of Congenital Myotonic Dystrophy. Neurobiol. Dis. 2019, 132, 104533.
- Harper, P.S. Myotonic Dystrophy, 3rd ed.; W.B. Saunders Company: London, UK; Philadelphia, PA, USA, 2001.
- López Castel, A.; Nakamori, M.; Tomé, S.; Chitayat, D.; Gourdon, G.; Thornton, C.A.; Pearson, C.E. Expanded CTG Repeat Demarcates a Boundary for Abnormal CpG Methylation in Myotonic Dystrophy Patient Tissues. Hum. Mol. Genet. 2011, 20, 1–15.
- Tomé, S.; Gourdon, G. DM1 Phenotype Variability and Triplet Repeat Instability: Challenges in the Development of New Therapies. Int. J. Mol. Sci. 2020, 21, 457.
- Harper, P.S.; Harley, H.G.; Reardon, W.; Shaw, D.J. Anticipation in Myotonic Dystrophy: New Light on an Old Problem. Am. J. Hum. Genet. 1992, 51, 10–16.
- Ashizawa, T.; Dunne, C.J.; Dubel, J.R.; Perryman, M.B.; Epstein, H.F.; Boerwinkle, E.; Hejtmancik, J.F. Anticipation in Myotonic Dystrophy: I. Statistical Verification Based on Clinical and Haplotype Findings. Neurology 1992, 42, 1871–1877.
- Dogan, C.; De Antonio, M.; Hamroun, D.; Varet, H.; Fabbro, M.; Rougier, F.; Amarof, K.; Arne Bes, M.-C.; Bedat-Millet, A.-L.; Behin, A.; et al. Gender as a Modifying Factor Influencing Myotonic Dystrophy Type 1 Phenotype Severity and Mortality: A Nationwide Multiple Databases Cross-Sectional Observational Study. PLoS ONE 2016, 11, e0148264.
- Garibaldi, M.; Lauletta, A.; Bucci, E.; Fionda, L.; Vanoli, F.; Leonardi, L.; Alfieri, G.; Tufano, L.; Morino, S.; Merlonghi, G.; et al. Gender Effect on Cardiac Involvement in Myotonic Dystrophy Type 1. Eur. J. Neurol. 2021, 28, 1366–1374.
- Barbé, L.; Lanni, S.; López-Castel, A.; Franck, S.; Spits, C.; Keymolen, K.; Seneca, S.; Tomé, S.; Miron, I.; Letourneau, J.; et al. CpG Methylation, a Parent-of-Origin Effect for Maternal-Biased Transmission of Congenital Myotonic Dystrophy. Am. J. Hum. Genet. 2017, 100, 488–505.
- Morales, F.; Corrales, E.; Zhang, B.; Vásquez, M.; Santamaría-Ulloa, C.; Quesada, H.; Sirito, M.; Estecio, M.R.; Monckton, D.G.; Krahe, R. Myotonic Dystrophy Type 1 (DM1) Clinical Subtypes and CTCF Site Methylation Status Flanking the CTG Expansion Are Mutant Allele Length-Dependent. Hum. Mol. Genet. 2021, 31, 262–274.
- Martorell, L.; Martinez, J.M.; Carey, N.; Johnson, K.; Baiget, M. Comparison of CTG Repeat Length Expansion and Clinical Progression of Myotonic Dystrophy over a Five Year Period. J. Med. Genet. 1995, 32, 593–596.
- Wong, L.J.; Ashizawa, T.; Monckton, D.G.; Caskey, C.T.; Richards, C.S. Somatic Heterogeneity of the CTG Repeat in Myotonic Dystrophy Is Age and Size Dependent. Am. J. Hum. Genet. 1995, 56, 114–122.
- Martorell, L.; Monckton, D.G.; Gamez, J.; Johnson, K.J.; Gich, I.; Lopez de Munain, A.; Baiget, M. Progression of Somatic CTG Repeat Length Heterogeneity in the Blood Cells of Myotonic Dystrophy Patients. Hum. Mol. Genet. 1998, 7, 307–312.
- Morales, F.; Vásquez, M.; Corrales, E.; Vindas-Smith, R.; Santamaría-Ulloa, C.; Zhang, B.; Sirito, M.; Estecio, M.R.; Krahe, R.; Monckton, D.G. Longitudinal Increases in Somatic Mosaicism of the Expanded CTG Repeat in Myotonic Dystrophy Type 1 Are Associated with Variation in Age-at-Onset. Hum. Mol. Genet. 2020, 29, 2496–2507.
- Martorell, L.; Johnson, K.; Boucher, C.A.; Baiget, M. Somatic Instability of the Myotonic Dystrophy (CTG)n Repeat during Human Fetal Development. Hum. Mol. Genet. 1997, 6, 877–880.
- Wöhrle, D.; Kennerknecht, I.; Wolf, M.; Enders, H.; Schwemmle, S.; Steinbach, P. Heterogeneity of DM Kinase Repeat Expansion in Different Fetal Tissues and Further Expansion during Cell Proliferation in Vitro: Evidence for a Casual Involvement of Methyl-Directed DNA Mismatch Repair in Triplet Repeat Stability. Hum. Mol. Genet. 1995, 4, 1147–1153.
- Kinoshita, M.; Takahashi, R.; Hasegawa, T.; Komori, T.; Nagasawa, R.; Hirose, K.; Tanabe, H. (CTG)n Expansions in Various Tissues from a Myotonic Dystrophy Patient. Muscle Nerve 1996, 19, 240–242.
- Cumming, S.A.; Jimenez-Moreno, C.; Okkersen, K.; Wenninger, S.; Daidj, F.; Hogarth, F.; Littleford, R.; Gorman, G.; Bassez, G.; Schoser, B.; et al. Genetic Determinants of Disease Severity in the Myotonic Dystrophy Type 1 OPTIMISTIC Cohort. Neurology 2019, 93, e995–e1009.
- Morales, F.; Couto, J.M.; Higham, C.F.; Hogg, G.; Cuenca, P.; Braida, C.; Wilson, R.H.; Adam, B.; del Valle, G.; Brian, R.; et al. Somatic Instability of the Expanded CTG Triplet Repeat in Myotonic Dystrophy Type 1 Is a Heritable Quantitative Trait and Modifier of Disease Severity. Hum. Mol. Genet. 2012, 21, 3558–3567.
- Overend, G.; Légaré, C.; Mathieu, J.; Bouchard, L.; Gagnon, C.; Monckton, D.G. Allele Length of the DMPK CTG Repeat Is a Predictor of Progressive Myotonic Dystrophy Type 1 Phenotypes. Hum. Mol. Genet. 2019, 28, 2245–2254.
- Botta, A.; Rossi, G.; Marcaurelio, M.; Fontana, L.; D’Apice, M.R.; Brancati, F.; Massa, R.; Monckton, D.G.; Sangiuolo, F.; Novelli, G. Identification and Characterization of 5’ CCG Interruptions in Complex DMPK Expanded Alleles. Eur. J. Hum. Genet. 2017, 25, 257–261.
- Braida, C.; Stefanatos, R.K.A.; Adam, B.; Mahajan, N.; Smeets, H.J.M.; Niel, F.; Goizet, C.; Arveiler, B.; Koenig, M.; Lagier-Tourenne, C.; et al. Variant CCG and GGC Repeats within the CTG Expansion Dramatically Modify Mutational Dynamics and Likely Contribute toward Unusual Symptoms in Some Myotonic Dystrophy Type 1 Patients. Hum. Mol. Genet. 2010, 19, 1399–1412.
- Cumming, S.A.; Hamilton, M.J.; Robb, Y.; Gregory, H.; McWilliam, C.; Cooper, A.; Adam, B.; McGhie, J.; Hamilton, G.; Herzyk, P.; et al. De Novo Repeat Interruptions Are Associated with Reduced Somatic Instability and Mild or Absent Clinical Features in Myotonic Dystrophy Type 1. Eur. J. Hum. Genet. 2018, 26, 1635–1647.
- Musova, Z.; Mazanec, R.; Krepelova, A.; Ehler, E.; Vales, J.; Jaklova, R.; Prochazka, T.; Koukal, P.; Marikova, T.; Kraus, J.; et al. Highly Unstable Sequence Interruptions of the CTG Repeat in the Myotonic Dystrophy Gene. Am. J. Med. Genet. A 2009, 149A, 1365–1374.
- Pešović, J.; Perić, S.; Brkušanin, M.; Brajušković, G.; Rakočević-Stojanović, V.; Savić-Pavićević, D. Molecular Genetic and Clinical Characterization of Myotonic Dystrophy Type 1 Patients Carrying Variant Repeats within DMPK Expansions. Neurogenetics 2017, 18, 207–218.
- Santoro, M.; Masciullo, M.; Silvestri, G.; Novelli, G.; Botta, A. Myotonic Dystrophy Type 1: Role of CCG, CTC and CGG Interruptions within DMPK Alleles in the Pathogenesis and Molecular Diagnosis. Clin. Genet. 2017, 92, 355–364.
- Tomé, S.; Dandelot, E.; Dogan, C.; Bertrand, A.; Geneviève, D.; Péréon, Y.; DM contraction study group; Simon, M.; Bonnefont, J.-P.; Bassez, G.; et al. Unusual Association of a Unique CAG Interruption in 5’ of DM1 CTG Repeats with Intergenerational Contractions and Low Somatic Mosaicism. Hum. Mutat. 2018, 39, 970–982.
- Miller, J.N.; van der Plas, E.; Hamilton, M.; Koscik, T.R.; Gutmann, L.; Cumming, S.A.; Monckton, D.G.; Nopoulos, P.C. Variant Repeats within the DMPK CTG Expansion Protect Function in Myotonic Dystrophy Type 1. Neurol. Genet. 2020, 6, e504.
- Wenninger, S.; Cumming, S.A.; Gutschmidt, K.; Okkersen, K.; Jimenez-Moreno, A.C.; Daidj, F.; Lochmüller, H.; Hogarth, F.; Knoop, H.; Bassez, G.; et al. Associations Between Variant Repeat Interruptions and Clinical Outcomes in Myotonic Dystrophy Type 1. Neurol. Genet. 2021, 7, e572.
- Pearson, C.E.; Nichol Edamura, K.; Cleary, J.D. Repeat Instability: Mechanisms of Dynamic Mutations. Nat. Rev. Genet. 2005, 6, 729–742.
- Foiry, L.; Dong, L.; Savouret, C.; Hubert, L.; te Riele, H.; Junien, C.; Gourdon, G. Msh3 Is a Limiting Factor in the Formation of Intergenerational CTG Expansions in DM1 Transgenic Mice. Hum. Genet. 2006, 119, 520–526.
- Savouret, C.; Brisson, E.; Essers, J.; Kanaar, R.; Pastink, A.; te Riele, H.; Junien, C.; Gourdon, G. CTG Repeat Instability and Size Variation Timing in DNA Repair-Deficient Mice. EMBO J. 2003, 22, 2264–2273.
- Flower, M.; Lomeikaite, V.; Ciosi, M.; Cumming, S.; Morales, F.; Lo, K.; Hensman Moss, D.; Jones, L.; Holmans, P.; TRACK-HD Investigators; et al. MSH3 Modifies Somatic Instability and Disease Severity in Huntington’s and Myotonic Dystrophy Type 1. Brain 2019, 142, 1876–1886.
- Morales, F.; Vásquez, M.; Santamaría, C.; Cuenca, P.; Corrales, E.; Monckton, D.G. A Polymorphism in the MSH3 Mismatch Repair Gene Is Associated with the Levels of Somatic Instability of the Expanded CTG Repeat in the Blood DNA of Myotonic Dystrophy Type 1 Patients. DNA Repair 2016, 40, 57–66.
- Breton, É.; Légaré, C.; Overend, G.; Guay, S.-P.; Monckton, D.; Mathieu, J.; Gagnon, C.; Richer, L.; Gallais, B.; Bouchard, L. DNA Methylation at the DMPK Gene Locus Is Associated with Cognitive Functions in Myotonic Dystrophy Type 1. Epigenomics 2020, 12, 2051–2064.
- Buckley, L.; Lacey, M.; Ehrlich, M. Epigenetics of the Myotonic Dystrophy-Associated DMPK Gene Neighborhood. Epigenomics 2016, 8, 13–31.
- Cho, D.H.; Thienes, C.P.; Mahoney, S.E.; Analau, E.; Filippova, G.N.; Tapscott, S.J. Antisense Transcription and Heterochromatin at the DM1 CTG Repeats Are Constrained by CTCF. Mol. Cell 2005, 20, 483–489.
- Filippova, G.N.; Thienes, C.P.; Penn, B.H.; Cho, D.H.; Hu, Y.J.; Moore, J.M.; Klesert, T.R.; Lobanenkov, V.V.; Tapscott, S.J. CTCF-Binding Sites Flank CTG/CAG Repeats and Form a Methylation-Sensitive Insulator at the DM1 Locus. Nat. Genet. 2001, 28, 335–343.
- Légaré, C.; Overend, G.; Guay, S.-P.; Monckton, D.G.; Mathieu, J.; Gagnon, C.; Bouchard, L. DMPK Gene DNA Methylation Levels Are Associated with Muscular and Respiratory Profiles in DM1. Neurol. Genet. 2019, 5, e338.
- Alwazzan, M.; Newman, E.; Hamshere, M.G.; Brook, J.D. Myotonic Dystrophy Is Associated with a Reduced Level of RNA from the DMWD Allele Adjacent to the Expanded Repeat. Hum. Mol. Genet. 1999, 8, 1491–1497.
- Eriksson, M. Simultaneous Analysis of Expression of the Three Myotonic Dystrophy Locus Genes in Adult Skeletal Muscle Samples: The CTG Expansion Correlates Inversely with DMPK and 59 Expression Levels, but Not DMAHP Levels. Hum. Mol. Genet. 1999, 8, 1053–1060.
- Frisch, R.; Singleton, K.R.; Moses, P.A.; Gonzalez, I.L.; Carango, P.; Marks, H.G.; Funanage, V.L. Effect of Triplet Repeat Expansion on Chromatin Structure and Expression of DMPK and Neighboring Genes, SIX5 and DMWD, in Myotonic Dystrophy. Mol. Genet. Metab. 2001, 74, 281–291.
- Klesert, T.R.; Otten, A.D.; Bird, T.D.; Tapscott, S.J. Trinucleotide Repeat Expansion at the Myotonic Dystrophy Locus Reduces Expression of DMAHP. Nat. Genet. 1997, 16, 402–406.
- Thornton, C.A.; Wymer, J.P.; Simmons, Z.; McClain, C.; Moxley, R.T. Expansion of the Myotonic Dystrophy CTG Repeat Reduces Expression of the Flanking DMAHP Gene. Nat. Genet. 1997, 16, 407–409.
- Kaliman, P.; Llagostera, E. Myotonic Dystrophy Protein Kinase (DMPK) and Its Role in the Pathogenesis of Myotonic Dystrophy 1. Cell. Signal. 2008, 20, 1935–1941.
- Klesert, T.R.; Cho, D.H.; Clark, J.I.; Maylie, J.; Adelman, J.; Snider, L.; Yuen, E.C.; Soriano, P.; Tapscott, S.J. Mice Deficient in Six5 Develop Cataracts: Implications for Myotonic Dystrophy. Nat. Genet. 2000, 25, 105–109.
- Krahe, R.; Ashizawa, T.; Abbruzzese, C.; Roeder, E.; Carango, P.; Giacanelli, M.; Funanage, V.L.; Siciliano, M.J. Effect of Myotonic Dystrophy Trinucleotide Repeat Expansion on DMPK Transcription and Processing. Genomics 1995, 28, 1–14.
- Sarkar, P.S.; Appukuttan, B.; Han, J.; Ito, Y.; Ai, C.; Tsai, W.; Chai, Y.; Stout, J.T.; Reddy, S. Heterozygous Loss of Six5 in Mice Is Sufficient to Cause Ocular Cataracts. Nat. Genet. 2000, 25, 110–114.
- Wakimoto, H.; Maguire, C.T.; Sherwood, M.C.; Vargas, M.M.; Sarkar, P.S.; Han, J.; Reddy, S.; Berul, C.I. Characterization of Cardiac Conduction System Abnormalities in Mice with Targeted Disruption of Six5 Gene. J. Interv. Card. Electrophysiol. 2002, 7, 127–135.
- Yin, Q.; Wang, H.; Li, N.; Ding, Y.; Xie, Z.; Jin, L.; Li, Y.; Wang, Q.; Liu, X.; Xu, L.; et al. Dosage Effect of Multiple Genes Accounts for Multisystem Disorder of Myotonic Dystrophy Type 1. Cell Res. 2020, 30, 133–145.
- Sicot, G.; Gomes-Pereira, M. RNA Toxicity in Human Disease and Animal Models: From the Uncovering of a New Mechanism to the Development of Promising Therapies. Biochim. Biophys. Acta 2013, 1832, 1390–1409.
- Wojciechowska, M.; Krzyzosiak, W.J. Cellular Toxicity of Expanded RNA Repeats: Focus on RNA Foci. Hum. Mol. Genet. 2011, 20, 3811–3821.
- Fugier, C.; Klein, A.F.; Hammer, C.; Vassilopoulos, S.; Ivarsson, Y.; Toussaint, A.; Tosch, V.; Vignaud, A.; Ferry, A.; Messaddeq, N.; et al. Misregulated Alternative Splicing of BIN1 Is Associated with T Tubule Alterations and Muscle Weakness in Myotonic Dystrophy. Nat. Med. 2011, 17, 720–725.
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A Muscleblind Knockout Model for Myotonic Dystrophy. Science 2003, 302, 1978–1980.
- Santoro, M.; Fontana, L.; Masciullo, M.; Bianchi, M.L.E.; Rossi, S.; Leoncini, E.; Novelli, G.; Botta, A.; Silvestri, G. Expansion Size and Presence of CCG/CTC/CGG Sequence Interruptions in the Expanded CTG Array Are Independently Associated to Hypermethylation at the DMPK Locus in Myotonic Dystrophy Type 1 (DM1). Biochim. Biophys. Acta 2015, 1852, 2645–2652.
- Steinbach, P.; Gläser, D.; Vogel, W.; Wolf, M.; Schwemmle, S. The DMPK Gene of Severely Affected Myotonic Dystrophy Patients Is Hypermethylated Proximal to the Largely Expanded CTG Repeat. Am. J. Hum. Genet. 1998, 62, 278–285.
- Perna, A.; Maccora, D.; Rossi, S.; Nicoletti, T.F.; Zocco, M.A.; Riso, V.; Modoni, A.; Petrucci, A.; Valenza, V.; Grieco, A.; et al. High Prevalence and Gender-Related Differences of Gastrointestinal Manifestations in a Cohort of DM1 Patients: A Perspective, Cross-Sectional Study. Front. Neurol. 2020, 11, 394.
- Hildonen, M.; Knak, K.L.; Dunø, M.; Vissing, J.; Tümer, Z. Stable Longitudinal Methylation Levels at the CpG Sites Flanking the CTG Repeat of DMPK in Patients with Myotonic Dystrophy Type 1. Genes 2020, 11, 936.
- Peric, S.; Pesovic, J.; Savic-Pavicevic, D.; Rakocevic Stojanovic, V.; Meola, G. Molecular and Clinical Implications of Variant Repeats in Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2021, 23, 354.
- Ballester-Lopez, A.; Koehorst, E.; Almendrote, M.; Martínez-Piñeiro, A.; Lucente, G.; Linares-Pardo, I.; Núñez-Manchón, J.; Guanyabens, N.; Cano, A.; Lucia, A.; et al. A DM1 Family with Interruptions Associated with Atypical Symptoms and Late Onset but Not with a Milder Phenotype. Hum. Mutat. 2020, 41, 420–431.
- Pešović, J.; Perić, S.; Brkušanin, M.; Brajušković, G.; Rakočević-Stojanović, V.; Savić-Pavićević, D. Repeat Interruptions Modify Age at Onset in Myotonic Dystrophy Type 1 by Stabilizing DMPK Expansions in Somatic Cells. Front. Genet. 2018, 9, 601.
- Brouwer, J.R.; Huguet, A.; Nicole, A.; Munnich, A.; Gourdon, G. Transcriptionally Repressive Chromatin Remodelling and CpG Methylation in the Presence of Expanded CTG-Repeats at the DM1 Locus. J. Nucleic Acids 2013, 2013, 567435.
- Otten, A.D.; Tapscott, S.J. Triplet Repeat Expansion in Myotonic Dystrophy Alters the Adjacent Chromatin Structure. Proc. Natl. Acad. Sci. USA 1995, 92, 5465–5469.
- Saveliev, A.; Everett, C.; Sharpe, T.; Webster, Z.; Festenstein, R. DNA Triplet Repeats Mediate Heterochromatin-Protein-1-Sensitive Variegated Gene Silencing. Nature 2003, 422, 909–913.
- Carrell, S.T.; Carrell, E.M.; Auerbach, D.; Pandey, S.K.; Bennett, C.F.; Dirksen, R.T.; Thornton, C.A. Dmpk Gene Deletion or Antisense Knockdown Does Not Compromise Cardiac or Skeletal Muscle Function in Mice. Hum. Mol. Genet. 2016, 25, 4328–4338.
- Appleton, G.O.; Li, Y.; Taffet, G.E.; Hartley, C.J.; Michael, L.H.; Entman, M.L.; Roberts, R.; Khoury, D.S. Determinants of Cardiac Electrophysiological Properties in Mice. J. Interv. Card. Electrophysiol. 2004, 11, 5–14.
- Chaves, A.A.; Dech, S.J.; Nakayama, T.; Hamlin, R.L.; Bauer, J.A.; Carnes, C.A. Age and Anesthetic Effects on Murine Electrocardiography. Life Sci. 2003, 72, 2401–2412.
- Müller, U. Ten Years of Gene Targeting: Targeted Mouse Mutants, from Vector Design to Phenotype Analysis. Mech. Dev. 1999, 82, 3–21.
- Gorbunova, V.; Seluanov, A.; Mittelman, D.; Wilson, J.H. Genome-Wide Demethylation Destabilizes CTG.CAG Trinucleotide Repeats in Mammalian Cells. Hum. Mol. Genet. 2004, 13, 2979–2989.
- Gomes-Pereira, M.; Monckton, D.G. Chemically Induced Increases and Decreases in the Rate of Expansion of a CAG * CTG Triplet Repeat. Nucleic Acids Res. 2004, 32, 2865–2872.
- Butler, R.; Bates, G.P. Histone Deacetylase Inhibitors as Therapeutics for Polyglutamine Disorders. Nat. Rev. Neurosci. 2006, 7, 784–796.
- Kazantsev, A.G.; Thompson, L.M. Therapeutic Application of Histone Deacetylase Inhibitors for Central Nervous System Disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868.
- Suelves, N.; Kirkham-McCarthy, L.; Lahue, R.S.; Ginés, S. A Selective Inhibitor of Histone Deacetylase 3 Prevents Cognitive Deficits and Suppresses Striatal CAG Repeat Expansions in Huntington’s Disease Mice. Sci. Rep. 2017, 7, 6082.
- Debacker, K.; Frizzell, A.; Gleeson, O.; Kirkham-McCarthy, L.; Mertz, T.; Lahue, R.S. Histone Deacetylase Complexes Promote Trinucleotide Repeat Expansions. PLoS Biol. 2012, 10, e1001257.
- Gannon, A.-M.M.; Frizzell, A.; Healy, E.; Lahue, R.S. MutSβ and Histone Deacetylase Complexes Promote Expansions of Trinucleotide Repeats in Human Cells. Nucleic Acids Res. 2012, 40, 10324–10333.
- van den Broek, W.J.A.A.; Nelen, M.R.; Wansink, D.G.; Coerwinkel, M.M.; te Riele, H.; Groenen, P.J.T.A.; Wieringa, B. Somatic Expansion Behaviour of the (CTG)n Repeat in Myotonic Dystrophy Knock-in Mice Is Differentially Affected by Msh3 and Msh6 Mismatch-Repair Proteins. Hum. Mol. Genet. 2002, 11, 191–198.
- Williams, G.M.; Paschalis, V.; Ortega, J.; Muskett, F.W.; Hodgkinson, J.T.; Li, G.-M.; Schwabe, J.W.R.; Lahue, R.S. HDAC3 Deacetylates the DNA Mismatch Repair Factor MutSβ to Stimulate Triplet Repeat Expansions. Proc. Natl. Acad. Sci. USA 2020, 117, 23597–23605.
- Ho, T.H.; Charlet-B, N.; Poulos, M.G.; Singh, G.; Swanson, M.S.; Cooper, T.A. Muscleblind Proteins Regulate Alternative Splicing. EMBO J. 2004, 23, 3103–3112.
- López-Martínez, A.; Soblechero-Martín, P.; de-la-Puente-Ovejero, L.; Nogales-Gadea, G.; Arechavala-Gomeza, V. An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes 2020, 11, 1109.
- Chen, G.; Masuda, A.; Konishi, H.; Ohkawara, B.; Ito, M.; Kinoshita, M.; Kiyama, H.; Matsuura, T.; Ohno, K. Phenylbutazone Induces Expression of MBNL1 and Suppresses Formation of MBNL1-CUG RNA Foci in a Mouse Model of Myotonic Dystrophy. Sci. Rep. 2016, 6, 25317.
- Kanadia, R.N.; Shin, J.; Yuan, Y.; Beattie, S.G.; Wheeler, T.M.; Thornton, C.A.; Swanson, M.S. Reversal of RNA Missplicing and Myotonia after Muscleblind Overexpression in a Mouse Poly(CUG) Model for Myotonic Dystrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 11748–11753.
- Huang, K.; Masuda, A.; Chen, G.; Bushra, S.; Kamon, M.; Araki, T.; Kinoshita, M.; Ohkawara, B.; Ito, M.; Ohno, K. Inhibition of Cyclooxygenase-1 by Nonsteroidal Anti-Inflammatory Drugs Demethylates MeR2 Enhancer and Promotes Mbnl1 Transcription in Myogenic Cells. Sci. Rep. 2020, 10, 2558.
- Li, S.; Tollefsbol, T.O. DNA Methylation Methods: Global DNA Methylation and Methylomic Analyses. Methods 2021, 187, 28–43.
- Hackett, J.A.; Sengupta, R.; Zylicz, J.J.; Murakami, K.; Lee, C.; Down, T.A.; Surani, M.A. Germline DNA Demethylation Dynamics and Imprint Erasure through 5-Hydroxymethylcytosine. Science 2013, 339, 448–452.
- Klug, M.; Schmidhofer, S.; Gebhard, C.; Andreesen, R.; Rehli, M. 5-Hydroxymethylcytosine Is an Essential Intermediate of Active DNA Demethylation Processes in Primary Human Monocytes. Genome Biol. 2013, 14, R46.
- Wu, H.; D’Alessio, A.C.; Ito, S.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Genome-Wide Analysis of 5-Hydroxymethylcytosine Distribution Reveals Its Dual Function in Transcriptional Regulation in Mouse Embryonic Stem Cells. Genes Dev. 2011, 25, 679–684.
- Colquitt, B.M.; Allen, W.E.; Barnea, G.; Lomvardas, S. Alteration of Genic 5-Hydroxymethylcytosine Patterning in Olfactory Neurons Correlates with Changes in Gene Expression and Cell Identity. Proc. Natl. Acad. Sci. USA 2013, 110, 14682–14687.
- Breton, C.V.; Salam, M.T.; Gilliland, F.D. Heritability and Role for the Environment in DNA Methylation in AXL Receptor Tyrosine Kinase. Epigenetics 2011, 6, 895–898.
- Hannon, E.; Knox, O.; Sugden, K.; Burrage, J.; Wong, C.C.Y.; Belsky, D.W.; Corcoran, D.L.; Arseneault, L.; Moffitt, T.E.; Caspi, A.; et al. Characterizing Genetic and Environmental Influences on Variable DNA Methylation Using Monozygotic and Dizygotic Twins. PLoS Genet. 2018, 14, e1007544.
- Waterland, R.A.; Jirtle, R.L. Early Nutrition, Epigenetic Changes at Transposons and Imprinted Genes, and Enhanced Susceptibility to Adult Chronic Diseases. Nutrition 2004, 20, 63–68.
- Xiao, F.-H.; Wang, H.-T.; Kong, Q.-P. Dynamic DNA Methylation During Aging: A “Prophet” of Age-Related Outcomes. Front. Genet. 2019, 10, 107.
- Gouil, Q.; Keniry, A. Latest Techniques to Study DNA Methylation. Essays Biochem. 2019, 63, 639–648.
- Liu, Y.; Rosikiewicz, W.; Pan, Z.; Jillette, N.; Wang, P.; Taghbalout, A.; Foox, J.; Mason, C.; Carroll, M.; Cheng, A.; et al. DNA Methylation-Calling Tools for Oxford Nanopore Sequencing: A Survey and Human Epigenome-Wide Evaluation. Genome Biol. 2021, 22, 295.
- Liu, Y.; Cheng, J.; Siejka-Zielińska, P.; Weldon, C.; Roberts, H.; Lopopolo, M.; Magri, A.; D’Arienzo, V.; Harris, J.M.; McKeating, J.A.; et al. Accurate Targeted Long-Read DNA Methylation and Hydroxymethylation Sequencing with TAPS. Genome Biol. 2020, 21, 54.
- Gigante, S.; Gouil, Q.; Lucattini, A.; Keniry, A.; Beck, T.; Tinning, M.; Gordon, L.; Woodruff, C.; Speed, T.P.; Blewitt, M.E.; et al. Using Long-Read Sequencing to Detect Imprinted DNA Methylation. Nucleic Acids Res. 2019, 47, e46.
- Biosciences, P. Detecting DNA Base Modifications Using Single Molecule, Real-Time Sequencing. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwji6pCaqIr2AhUC1hoKHRJQD0cQFnoECCwQAQ&url=https%3A%2F%2Fwww.pacb.com%2Fwp-content%2Fuploads%2F2015%2F09%2FWP_Detecting_DNA_Base_Modifications_Using_SMRT_Sequencing.pdf&usg=AOvVaw2HWFCF7I6LWvvQeU-50aru (accessed on 18 February 2022).
- Laszlo, A.H.; Derrington, I.M.; Brinkerhoff, H.; Langford, K.W.; Nova, I.C.; Samson, J.M.; Bartlett, J.J.; Pavlenok, M.; Gundlach, J.H. Detection and Mapping of 5-Methylcytosine and 5-Hydroxymethylcytosine with Nanopore MspA. Proc. Natl. Acad. Sci. USA 2013, 110, 18904–18909.
- Schreiber, J.; Wescoe, Z.L.; Abu-Shumays, R.; Vivian, J.T.; Baatar, B.; Karplus, K.; Akeson, M. Error Rates for Nanopore Discrimination among Cytosine, Methylcytosine, and Hydroxymethylcytosine along Individual DNA Strands. Proc. Natl. Acad. Sci. USA 2013, 110, 18910–18915.

