Among the trinucleotide repeat disorders, myotonic dystrophy type 1 (DM1) is one of the most complex neuromuscular diseases caused by an unstable CTG repeat expansion in the DMPK gene. DM1 patients exhibit high variability in the dynamics of CTG repeat instability and in the manifestations and progression of the disease. The largest expanded alleles are generally associated with the earliest and most severe clinical form. However, CTG repeat length alone is not sufficient to predict disease severity and progression, suggesting the involvement of other factors. Several data support the role of epigenetic alterations in clinical and genetic variability observed in DM1.
- myotonic dystrophy type 1 (DM1)
- DM1 phenotype
- CTG repeat expansion
- DNA methylation and chromatin
1. Introduction


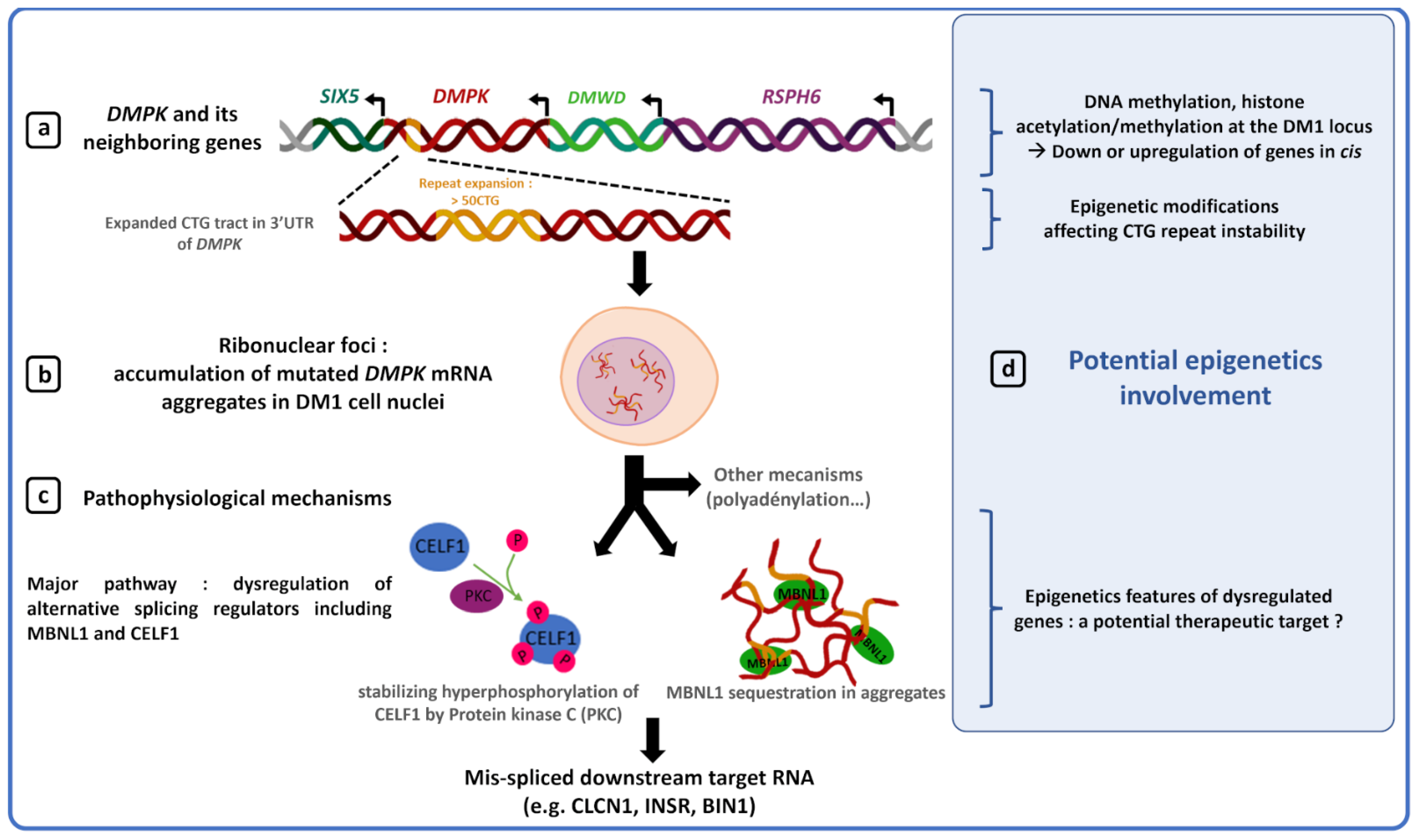
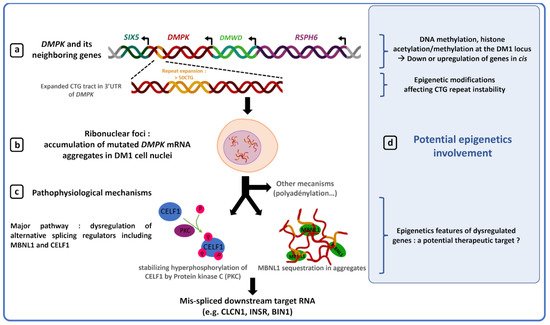
2. Epigenetic Modifications and DM1 Clinical Features
2.1. CDM1 Form Associated with Hypermethylation of the DM1 Locus and Large CTG Repeat Expansions
2.2. DM1 Symptoms Associated with Specific Epigenetic Patterns
DM1 is a multisystemic disease that affects all organs with high clinical variability resulting in great difficulty to predict the different symptoms and their progression. Two recent studies quantified methylation status of 57 CpG sites at the DMPK locus in blood of 90 and 115 DM1 patients with adult or late-onset forms using the bisulfite pyrosequencing method [44][48][44,48]. Légaré et al. showed that DNA methylation at two CpG sites downstream of the CTG repeat expansion correlates with respiratory failure and muscle weakness, independently of the CTG repeat length [48]. Similarly, a longitudinal study conducted over 9 years of follow-up in blood of 115 DM1 patients with adult form suggested that DNA methylation levels downstream of the CTG repeat were associated with changes in cognitive features [44]. In these two studies, the association between methylation at the DMPK locus and muscle, respiratory or cognitive phenotypes is highly dependent on methylation pattern of specific CpG sites.3. Sex, CTG Repeats and Epigenetics
3.1. DNA Methylation at the
DMPK
Locus May Contribute to Sex Differences in DM1 Symptoms
3.2. DNA Methylation Alters the Dynamics of CTG Repeat Instability- Role in the DM1 Phenotype?
DM1 patients showed expansion-biased intergenrational instability, providing a molecular explanation for the anticipation phenomenon defined by increased severity and earlier age of onset over generations [14]. The dynamics of instability depends on the sex and length of the CTG repeats of the transmitting parents. Larger expansions are often maternally transmitted, which explain the maternal transmission bias of CDM1. Recent studies have described that upstream methylation at the DM1 locus was mainly found in patients carrying a maternally inherited expanded allele (>500 CTG) in which epigenetics modification could be responsible for the discrepancy in CDM1 transmission risk between maternal and paternal transmissions [18][19][64][67][18,19,64,85]. Somatic mosaicism also contributes to the progressive nature of the disease in DM1 [27][28][27, 28]. DM1 patients with the most rapid increase in triplet size over time in the blood develop earlier and more severe symptoms [23][27][28][23, 27, 28]. Furthermore, an increase in somatic mosaicism in the blood of the same individual over time is also associated with a change in the age of onset [23]. DNA methylation may contribute to disease development and progression by modulating the dynamics of CTG repeat instability in DM1 patient tissues, either through methylation changes at the DM1 locus or elsewhere in the genome by altering the activity of genes involved in CTG repeat instability.3.3. DM1 Locus Methylation Deregulated in Patients with an Interrupted Expanded Allele and Less Severe Symptoms
The majority of DM1 patients inherit a pure expansion of CTG repeats. However, 4–9% of the DM1 population carries CTG repeat interruptions, which differ in number and/or type across generations and tissues [27][29][30][31][32][33][34][35][36][37][38][68][69][70][27,29,30,31,32,33,34,35,36,37,38,87,88,89]. Repeat interruptions are associated with stabilizations and/or contractions of the CTG repeat expansion and generally milder symptoms [31][32][36][37][64][68][69][70][31,32,36,37,64,87,88,89]. The mechanisms by which interrupted expanded alleles stabilize the repeat and then decrease the severity of DM1 symptoms in patients remain poorly understood. DNA methylation in the vicinity of the CTG repeats has been studied in DM1 patients carrying an interrupted expanded allele compared to DM1 patient with a pure CTG repeat tract. By bisulfite sequencing, no difference in CpG methylation pattern between two patients with 150 and 300 interrupted CTG repeats and matched DM1 patients with a pure repeat was found [36]. However, Santoro et al. showed a partially reversed methylation profile compared to pure repeat: increased CpG methylation downstream but no upstream CpG methylation in DM1 patients carrying 400–-900 interrupted CTG repeats [64]. Similarly, increased downstream methylation have also been found in other studies in patients carrying interrupted repeats [44][48][67][44,48,85]. Taken together, these observations suggest that interruptions could cause a change in the CpG methylation profile. However, it is difficult to conclude whether this change in methylation profile plays a role in the lower level of somatic instability of interrupted CTG repeats and milder DM1 symptoms or whether it is only a consequence.4. Epigenetics, Gene Expression and Chromatin at the DM1 Locus
Several studies have shown that abnormal CTG repeat expansion promotes DNA hypermethylation and heterochromatin formation at the DM1 locus, making this locus less accessible to transcription factors and eventually causing downregulation of DMPK and its neighboring genes, SIX 5 and DMWD [47][49][50][51][52][53][71][72][73][47,49,50,51,52,53,90,91,92]. Although decreased gene expression at the DM1 locus is not sufficient to explain the complex DM1 phenotype, downregulation of DMPK, SIX5 and DMWD genes by epigenic changes in DM1 could participate in some clinical features such as DM1 cataracts, muscle defects or cardiac conduction defects. Indeed, generation of mouse models has shown that decreased DMPK expression induces late myopathy and mild cardiac conduction defects [54]. However, recently, the generation of a novel dmpk knockout mouse model did not reveal any significant defects in cardiac and skeletal muscles [74][93]. Such a difference between studies could be explained by different parameters such as a different genetic background between models or a difference in model characterization techniques. Earliest studies performed heavy anesthesia on mice while it is now known to disrupt the ECG [75][76][94,95]. Furthermore, selection cassettes, used to generate some knockout dmpk mouse models, are known to possibly interfere with the expression of flanking genes [77][96]. Haploinsufficiency of the six5 gene induces cataracts with incomplete penetrance as well as mild cardiac conduction defects [55][57][58][55,57,58]. In a dmwd+/− mice, a dramatic reduction of myofiber cross-sectional area was observed, suggesting a potential role of DMWD dysregulation in the DM1 muscular phenotype [59]. The chromatin structure and methylation of the DM1 locus could participate in the pathogenesis of DM1 by reducing expression of genes at the DM1 locus, but, to date, no study directly prove this hypothesis.5. Epigenetic and Therapy for DM1
5.1. Modulation of Somatic Mosaicism by Global Demethylation in DM1 Models
5.2. Modulation of CTG Somatic Instability by Inhibition of Histone Deacetylase
The function of histone deacetylases (HDACs) is to remove acetyl groups from histones and other proteins, which has functional consequences on chromatin structure and gene expression profiles. Several studies have shown the efficacy of HDAC inhibitors in trinucleotide expansion diseases such as polyglutamine diseases [80][81][82][111,112,113]. More recently, it was shown that inhibition of HDAC3 or HDAC5 with small molecules or siRNAs induced a significant reduction in expansion frequency in modified human astrocyte cell lines containing CTG repeats [83][84][114,115].In the same model, Williams etet al. al. demonstrated that RGFP966, a selective HDAC3 inhibitor, affects repeat instability by deacetylating MSH3 protein, a protein involved in the formation of repeat expansions in DM1 mouse models [40][85][86][40,116,117]. Inhibition of HDACs could contribute to attenuate DM1 disease worsening by modulating repeat instability or another pathway that needs to be identified. However, further studies in murine models are needed to ensure that the use of HDAC inhibitors is truly beneficial to the patient without inducing deleterious side effects.
5.3. Modulation of MBNL1 Transcription by a Methyl Sensitive Enhancer
In DM1, toxic DMPK RNAs with CUG expansion induce a sequestration and inhibition of the RNA-binding protein MBNL1 that contributes to aberrant splicing providing an explanation for many of the symptoms observed in DM1 patients [62][87][88][62,118,119]. Previous studies have shown that increasing MBNL1 proteins or mRNA levels by a minimum 1.5-fold were sufficient to partially reverse aberrant splicing and ameliorate the phenotype of DM1 mice [89][90][120,121]. Recently, it was shown that demethylation of MeR2, a transcriptional enhancer located in the intron 1 of MBNL1 MBNL1, promotes Mbnl1 Mbnl1 transcription in C2C12 myoblasts as well as in mouse skeletal muscles [89][91][120, 122]. Thus, acting on the epigenetic processes to increase MBNL1 MBNL1 transcription and restore free MBNL1 protein level, may be considered as a potential new therapeutic target to restore normal splicing and reduce DM1 symptoms.6. Conclusions
DNA methylation and chromatin structure are two altered parameters in DM1 with an overrepresentation of studies analyzing the methylation of CpG sites around CTG repeats [12][18][19][23][47][51][64][65][72][73][12,18,19,23,47,51,64,65,91,92]. Some data remain controversial and result from the variability observed between methods, regions analyzed and genetic and clinical description of patients. Despite this observed heterogeneity between studies, most studies show that the region upstream of CTG repeats is generally hypermethylated in CDM1 patients compared to unaffected individuals and patients with less severe forms [12][18][19][64][65][67][12,18,19,64,65,85]. To date, the use of methylation alone as a biomarker seems difficult without estimation of the size of the inherited allele by a state-of-the-art method in patients. However, large CTG expansions associated with DNA hypermethylation around the expanded CTG repeat are strong indicators in favor of CDM1. These two indicators would be a powerful tool to predict the most severe forms of DM1 and improve prenatal diagnosis and genetic counseling. Moreover, two recent papers have associated hypermethylation of CpG sites at the DM1 locus with symptoms affecting respiratory, muscular and cognitive functions. These data suggest that accurate mapping of CpG methylation would be necessary to improve prognosis and thus propose earlier treatments to patients to limit disease worsening [44][48][44,48]. Interestingly, DNA hydroxymethylation has not been studied in DM1 patients. Recently, aberrant DNA hydroxymethylation changes have been observed in cancers and neurological diseases such as Alzheimer’s disease suggesting an important role of the epigenomic mark hydroxymethylcytosine (5hmC) in tumorigenesis and the neuronal system [92][123]. 5hmC plays a key role in different pathways such as initiation of demethylation and gene expression regulation in mammals [93][94][95][96][124,125,126,127]. Analysis of DNA hydroxymethylation at the DM1 locus may provide a new insight into the function of epigenetic modifications in the dynamics of repeat instability but also in the pathogenesis of DM1.Although promising, the use of methylation as biomarker to refine prognosis and improve treatments must be taken with caution as various external and internal factors such as the environment or the patient’'s age at the time of sampling are known to affect methylation levels [11, [11][23][97][98][99][100]23,128, 129, 130, 131]. Chromatin structure and gene expression modulated by epigenetic processes are less studied in DM1. It is not yet known whether these parameters could be used as biomarkers in DM1. However, they remain interesting potential therapeutic targets to alleviate DM1 symptoms by epigenetic regulation of activator or repressor of dysfunctional DM1 proteins (MBNL1) or reduction of somatic instability (HDAC inhibitors) [83][84][86][89][91][114,115,117,120,122].
Analyses of epigenetic modifications are not straightforward and require homogenization of data through better characterization of the DM1 mutation, DM1 clinics and methylation status at the DM1 locus. The emergence of long-read sequencing technologies in DM1 offers new perspectives to characterize DM1 expansion but also methylation (DNA methylation and hydroxymethylation) at the DM1 locus without DNA amplification. Single-molecule real-time (SMRT) sequencing from Pacific Biosciences and nanopore sequencing by Oxford Nanopore Technologies are the two long-read sequencing technologies capable of detecting DNA base methylation [101][102][103][67,68,69]. Both methods can discriminate information between parental alleles, which is important to precisely characterize the CpG methylation pattern of the expanded CTG tract [104][132]. The two systems rely on very different technologies to detect CpG methylation on long reads. SMRT sequencing technology detects CpG methylation through subtle changes in polymerase kinetics throughout DNA synthesis [105][133]. On the other hand, nanopore sequencing technology directly detects CpG methylation via the difference produced in the electric current intensity between an unmodified and a methylated base upon passage through the pores [106][107][134,135]. Use of these two methods, PacBio and Oxford nanopore sequencing, to determine the exact CpG methylation at the DM1 locus, quantify the number of repeats, identify CNG interruptions, and estimate somatic mosaicism could provide new insights into the understanding of DM1 pathogenesis and allow for the development of more personalized treatment for DM1 patients.