Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Asparuh Nikolov | -- | 1689 | 2022-04-06 15:14:14 | | | |
| 2 | Rita Xu | Meta information modification | 1689 | 2022-04-07 05:09:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nikolov, A.; Popovski, N. Extracellular Matrix in Heart Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/21417 (accessed on 24 July 2026).
Nikolov A, Popovski N. Extracellular Matrix in Heart Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/21417. Accessed July 24, 2026.
Nikolov, Asparuh, Nikola Popovski. "Extracellular Matrix in Heart Disease" Encyclopedia, https://encyclopedia.pub/entry/21417 (accessed July 24, 2026).
Nikolov, A., & Popovski, N. (2022, April 06). Extracellular Matrix in Heart Disease. In Encyclopedia. https://encyclopedia.pub/entry/21417
Nikolov, Asparuh and Nikola Popovski. "Extracellular Matrix in Heart Disease." Encyclopedia. Web. 06 April, 2022.
Copy Citation
Accumulating evidence indicates that two major proteins are responsible for the structural coherence of bounding cardiomyocytes. These biomolecules are known as myocardial fibrillar collagen type I (COL1) and type III (COL3). In addition, fibronectin, laminin, fibrillin, elastin, glycoproteins, and proteoglycans take part in the formation of cardiac extracellular matrix (ECM). In physiological conditions, collagen synthesis and degradation in human cardiac ECM are well-regulated processes, but they can be impaired in certain cardiovascular diseases, such as heart failure (HF). Myocardial remodeling is part of the central mechanism of HF and involves cardiomyocyte injury and cardiac fibrosis due to increased fibrillar collagen accumulation.
extracellular matrix
collagen type I and III derived peptides
heart failure
1. Introduction
Heart failure (HF) has been recognized as a worldwide health burden that affects approximately 40 million people globally [1]. It has been estimated that the incidence of HF in adults is about 2%, and the rate rises to 6–10% over the age of 65 [2]. For those older than 75 years, the rate is more than 10% [3]. In addition, because of the increased life expectancy and risk factors such as hypertension, diabetes, dyslipidemia, and obesity, the morbidity rate is also expected to rise [4]. It has been reported that in people over the age of 65, heart failure is the leading cause of hospitalization.
Based on left ventricular ejection fraction (LVEF) values, the European Society of Cardiology (ESC) divides HF into three types: with preserved ejection fraction (HFpEF), characterized by LVEF ≥ 50%; mid-range (HFmrEF), with LVEF of 40–49%; and with reduced ejection fraction (HFrEF), with LVEF < 40% [5]. Considering the underlying etiologies, demographics, comorbidities, and responses to therapy, differentiation of HF according to LVEF has significant practical value.
2. Type I and Type III Collagen Characteristics
Collagen (COL) is the main fibrous protein in human ECM, accounting for more than one-third of total protein content in the organism [6]. Practically, it is present in all body systems containing connective tissue. Collagen is responsible for the strength and stability of the cytoskeleton and regulates normal cell and tissue development [7]. Different COL types form collagen fibers, so they represent a heterogeneous mix. However, in any given tissue, a certain type of collagen usually prevails [8].
Collagen type I is a fibrillar protein that makes up a large part of the structure of the interstitial membrane. It is known as the most common type of collagen, and is an important structural component of many tissues. COL1 can be found in almost all connective tissue structures. It is a structural protein found in bones; skin, tendons; ligaments; sclera; corneas; and blood vessels, as well as other tissues. It is a component aligned in fibers, thus forming a structural-mechanical scaffold (matrix) for bones; skin; tendons; corneas; blood vessel walls; and other connective tissues. The dominant isoform of COL1 is heterotrimers with two α1 (I) and one α2 (I) chain. In fetal tissues and some fibrous lesions, homotrimers with three α1 (I) chains have been discovered [9]. The homotrimeric isoform is known to be less susceptible to cleavage by collagenases, which may clarify its accumulation and functional role in tumors and fibrotic lesions [10].
Collagen type III has a unique molecular structure. A long protein chain is responsible for its tensile stiffness and the biomechanical characteristics of tissues. This contributes to the specific ECM properties when this type of collagen predominates. It is an important component of reticular fibers in the interstitial tissue of the lungs, liver, heart, and vessels [11].
Collagen type III is made up of only one collagen α chain. COL3 is a homotrimer made up of three α1 (III) chains overlapped in a right triple helix. It is produced by fibroblasts and other mesenchymal cells and plays an important role in inflammatory conditions such as lung damage, liver diseases, and renal and vascular fibrosis. Consequently, COL3 and COL1 are both important components of the myocardial ECM [12]. Today, immunological markers based on collagen type I and III turnover have been extensively investigated for detection of cardiac fibrosis.
3. Cardiac Extracellular Matrix: Structure and Function
The extracellular matrix (ECM) is made up of a fibrillar network along with a basement membrane, proteoglycans, and fibrous proteins such as fibronectins, collagens, elastins, fibrillins, and laminins [13]. Together they maintain the structural coherence of bounding cells, ensuring stability. The ECM has also been associated with the transmission of important biochemical signals, which are crucial for normal tissue development. ECM is present in all tissues, but each organ has a unique distribution of matrix components [14]. For example, cardiac ECM is primarily composed of collagen type I (85%) and III (11%). The traditional concept of myocardial ECM was that it was an inert mechanical scaffold providing structural cardiac integrity. Nowadays, it is considered as a dynamic network with important metabolic activity and many complex functions, such as regulation of molecular signaling; cell proliferation; differentiation, migration; adhesion; and protein interactions. Moreover, it also regulates myocardial remodeling in normal and pathological conditions. Therefore, cardiac ECM plays a fundamental role in maintaining cardiovascular homeostasis [15]. With the accumulation of additional knowledge about the structure and function of heart ECM, cardiac fibroblasts have been described as the primary source of myocardial COL1 and COL3 peptides. It can be concluded that they are the main cells producing collagen in the heart [14][15].
Cardiac fibroblasts are the major heart cells producing COL1 and COL3. Fibrillar collagen is initially synthetized as a procollagen, which is then split by specific proteinases into carboxy (C)- and amino (N)-terminal propeptides: N-terminal propeptides of COL1 and COL3 (PINP and PIIINP) and C-terminal propeptides (PICP and PIIICP). Thereafter, they are secreted in the circulation. Since propeptides are split, the triple helix chain “will form big collagen fibers with other collagen chains” [16]. Collagenases MMP-1, -8, and -13 degrade these collagen fibers, and telopeptides are formed during this process. Then the small telopeptides of collagen type I (ICTP, 12 kDa) are released into the circulation [17]. The big telopeptides go through spontaneous denaturation in nonhelical derivatives [18]. Subsequently, gelatinases MMP-2 and -9 completely degrade them into inactive fragments (Figure 1).
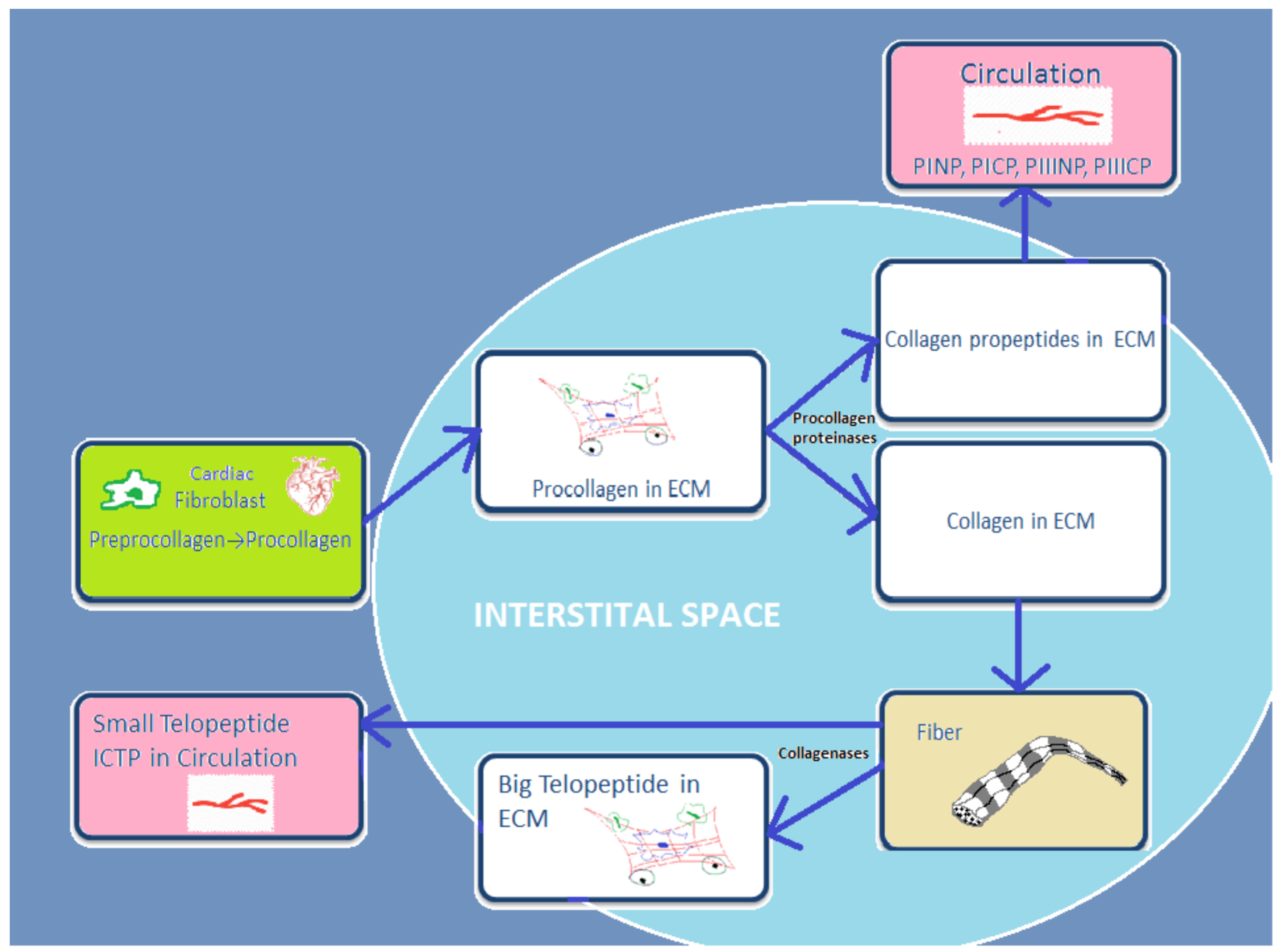
Figure 1. Schematic presentation of basic stages from process of synthesis and degradation of collagen I and III.
4. General Concepts of Abnormal Cardiac Extracellular Matrix Changes in Heart Failure
ECM is a dynamic structure that plays a crucial role in the development and progression of many cardiovascular diseases. Accumulating data indicate that fibrosis is observed in different cardiovascular diseases (CVDs). HF is an example of abnormal collagen accumulation, which pathologically increases myocardial stiffness and impairs heart contractile properties. Several CVDs, including hypertension; coronary artery disease; valvular disease; and arrhythmias, are considered to be leading causes of HF. A link has been found between cardiac remodeling and the development of HF [19]. Cardiac remodeling is defined as “a group of molecular, cellular and interstitial changes that manifest clinically as alterations in the size, mass, geometry and function of the heart after a stressful stimulus.” [20][21]. This process can be triggered by “ischemia (myocardial infarction), inflammation (myocarditis), hemodynamic overload (workload by volume or pressure) and neurohormonal activation” [22][23][24].
Paradoxically, cardiac remodeling is thought to be both an adaptive and a maladaptive process. Initially, cellular changes in the heart structure, such as myocyte hypertrophy, necrosis, and apoptosis, occur and then extracellular matrix deposition of fibrillar collagen increases (a process often defined as “myocardial fibrosis”) [25][26][27]. This has been related to impaired collagen metabolism, manifesting as accelerated synthesis and accumulation of COL1 and COL3 in the myocardium [28][29]. Accordingly, collagen degradation is slowed down and heart function is inevitably altered in the later stages of cardiac remodeling [30].
5. Basic Underlying Mechanisms of Myocardial Fibrosis in Heart Failure: Role of Impaired Type I and III Collagen Turnover
There is a large body of evidence regarding the accelerated myocardial accumulation of fibrillar collagen in heart failure. Early research on HF showed that various MMPs are present in the myocardium of patients with chronic heart failure [31]; since then, there has been ongoing enthusiasm for the routine application of COL1 and COL3 as biological markers for assessing cardiac tissue remodeling and myocardial fibrosis. This is true for both laboratory models and clinical studies [32]. In important experiments, Douglas et al.’s [31] and Alla’s [32] findings suggested delayed degradation of collagen in patients with chronic HF, thereby contributing to the mechanism of myocardial fibrosis development.
Increased serum levels of COL1 and COL3 synthesis biomarkers (PICP, PINP, PIIINCP, PIIINP) and decreased serum levels of the COL1 degradation biomarker (ICTP) have been linked to myocardial collagen deposition and fibrosis [33][34][35]. According to these findings, the equilibrium between synthesis and degradation of cardiac collagen is disrupted in heart disease [31][32]. Importantly, heart failure is an example of CVD disease that presents with altered collagen turnover [36][37].
A widely held concept regarding the pathogenesis of myocardial fibrosis is that cardiac lesion is considered to be an initiating event [38]. Thus, the MMP/TIMP system fails and the degradation activity of MMP-1, -2, -8, -9, and -13 is disturbed. As a result, fibroblasts in the heart are hyperactivated and transdifferentiated into myofibroblasts, which increase the production of collagen types I and III, then degradation processes decrease and abnormal collagen deposition in myocardium occurs [39][40]. Therefore, impaired collagen turnover abnormally affects the remodeling of cardiac ECM, and COL1-/COL3-derived peptides are released into the circulation. This can trigger an incessant vicious cycle of COL1/COL3 over-deposition and consequent suppressed degradation.All of these processes might contribute tothe development of cardiac fibrosis in heart failure. However, the molecular mechanisms of the genesis and progression of myocardial fibrosis are not yet fully clear (Figure 2) [39][40][41][42][43][44][45].
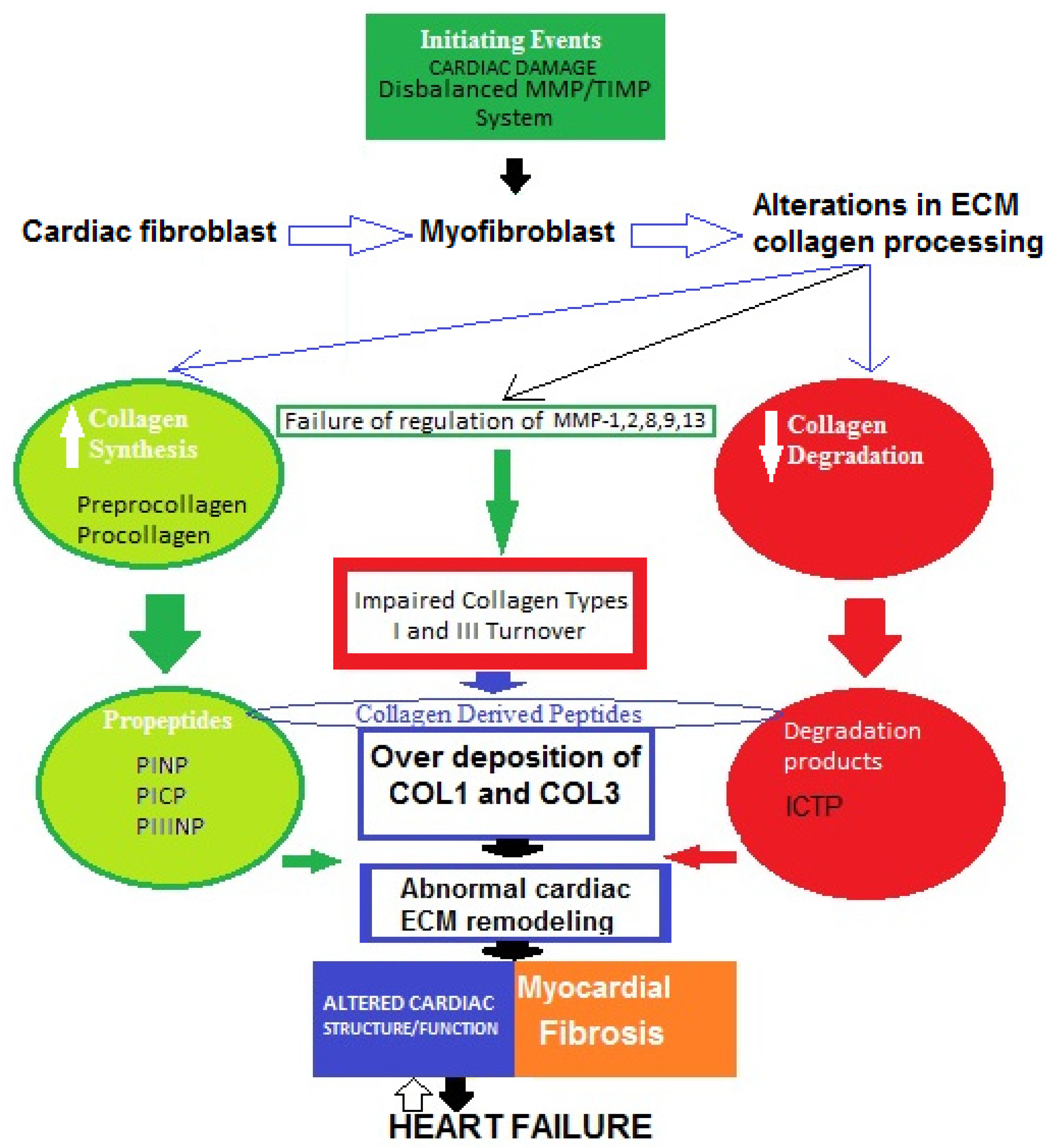
Figure 2. Possible schematic pattern illustrating eventual mechanisms of impaired collagen I and III turnover leading to myocardial fibrosis in heart failure.
The four major collagen-derived peptides of research interest nowadays are the N-terminal propeptide of collagen type I (PINP), the N-terminal propeptide of collagen type III (PIIINP), the C-terminal propeptide of collagen type I (PICP), and the C-terminal telopeptide of collagen type I (ICTP). PINP and PICP indicate collagen type I synthesis, PIIINP reflects collagen type III synthesis, and ICTP represents collagen type I degradation. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs (TIMPs) are other molecules produced by cardiac fibroblasts. MMPs are proteolytic enzymes that degrade ECM proteins and TIMPs are MMP inhibitors, maintaining a fine balance between synthesis and degradation processes. Thus, both MMPs and TIMPs are regulatory proteins essential for ECM homeostasis.
References
- GBD 2015. Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study. Lancet 2015, 388, 10053.
- Dickstein, K.; Cohen-Solal, A.; Filippatos, G.; McMurray, J.J.; Ponikowski, P.; Poole-Wilson, P.A. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur. Heart J. 2018, 29, 2388–2442.
- Metra, M.; Teerlink, J.R. Heart failure. Lancet 2017, 390, 1981–1995.
- Mann, D.L.; Chakinala, M. Harrison’s Principles of Internal Medicine: Heart Failure and Cor Pulmonale, 18th ed.; McGraw-Hill: New York, NY, USA, 2012; Chapter 234; pp. 1–11.
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 3599–3726.
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140.
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238.
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200.
- Henriksen, K.; Karsdal, M.A. Type I Collagen. In Biochemistry of Collagens, Laminins and Elastin Structure, Function and Biomarkers, 1st ed.; Karsdal, M.A., Ed.; Academic Press: Cambridge, MA, USA, 2016; Chapter 1; pp. 1–11.
- Han, S.; Makareeva, E.; Kuznetsova, N.V. Molecular mechanism of type I collagen homotrimer resistance to mammalian collagenases. J. Biol. Chem. 2010, 285, 22276–22281.
- Von Der Mark, K. Localization of collagen types in tissues. Int. Rev. Connect. Tissue Res. 1981, 9, 265–324.
- Nielsen, M.J.; Karsdal, M.A. Type III Collagen. In Biochemistry of Collagens, Laminins and Elastin Structure, Function and Biomarkers, 1st ed.; Karsdal, M.A., Ed.; Academic Press: Cambridge, MA, USA, 2016; Chapter 3; pp. 21–30.
- Jarvelainen, H.; Sainio, A.; Koulu, M.; Wight, T.N.; Penttinen, R. Extracellular matrix molecules: Potential targets in pharmacotherapy. Pharmacol. Rev. 2009, 61, 198–223.
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: London, UK, 2007.
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574.
- Anderson, K.R.; Sutton, M.G.; Lie, J.T. Histopathological types of cardiac fibrosis in myocardial disease. J. Pathol. 1978, 128, 79–85.
- Eghbali, M.; Czaja, M.J.; Zeydel, M.; Weiner, F.; Zern, M.A.; Seifter, S.; Blumenfefd, O.O. Colllagen chain mRNAs in isolated heart cells from young and adult rats. J. Mol. Cell. Cardiol. 1988, 20, 267–276.
- Lijnen, P.; Petrov, V.; Fagard, R. Induction of cardiac fibrosis by transforming growth factor-β1. Mol. Gen. Metab. 2000, 71, 418–435.
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling-concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582.
- Hockman, J.S.; Bulkley, B.H. Expansion of acute myocardial infarction: An experimental study. Circulation 1982, 65, 1446–1450.
- Pfeffer, M.A.; Braunwald, E. Ventricular remodeling after myocardial infarction: Experimental observations and clinical implications. Circulation 1990, 81, 1161–1172.
- Gaasch, W.H. Left ventricular radius to wall thickness ratio. Am. J. Cardiol. 1979, 43, 1189–1194.
- Sayer, G.; Bhat, G. The renin-angiotensin-aldosterone system and heart failure. Cardiol. Clin. 2014, 32, 21–32.
- Florea, V.G.; Cohn, J.N. The autonomic nervous system and heart failure. Circ. Res. 2014, 114, 1815–1826.
- Tan, L.B.; Jalil, J.E.; Pick, R. Cardiac myocyte necrosis induced by angiotensin II. Circ. Res. 1991, 69, 1185–1195.
- Sharov, V.G.; Sabbah, H.N.; Shimoyama, H. Evidence of cardiocyte apoptosis in myocardium of dogs with chronic heart failure. Am. J. Pathol. 1996, 148, 141–149.
- Teiger, E.; Dam, T.V.; Richard, L. Apoptosis in pressure overload—Induced heart hypertrophy in the rat. J. Clin. Investig. 1996, 97, 2891–2897.
- Olivetti, G.; Abbi, R.; Quaini, F. Apoptosis in the failing human heart. N. Engl. J. Med. 1997, 336, 1131–1141.
- Villarreal, F.J.; Kim, N.N.; Ungab, G.D. Identification of functional angiotensin II receptors on rat cardiac fibroblastos. Circulation 1993, 88, 2849–2861.
- Weber, K.T.; Pick, R.; Silver, M.A. Fibrillar collagen and remodeling of dilated canine left ventricle. Circulation 1990, 82, 1387–1401.
- Douglas, L.; Mann, M.D.; Spinale, F.G. Activation of Matrix Metalloproteinases in the Failing Human Heart Breaking the Tie That Binds. Circulation 1998, 98, 1699–1702.
- Alla, F. Early changes in serum markers of cardiac extra-cellular matrix turnover in patients with uncomplicated hypertension and type II diabetes. Eur. J. Heart Fail. 2006, 8, 147–153.
- Diez, J.; Querejeta, R.; Lopez, B. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation 2002, 105, 2512–2517.
- Laviades, C.; Varo, N.; Fernandez, J. Abnormalities of the extracellular degradation of collagen type I in essential hypertension. Circulation 1998, 98, 535–540.
- Fan, D.; Takawale, A.; Lee, J. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 5–15.
- Eghbali, M. Cardiac fibroblasts: Function, regulation of gene expression, and phenotypic modulation. Basic Res. Cardiol. 1992, 87 (Suppl. S2), 183–189.
- Moore, L.; Fan, D.; Basu, R. Tissue inhibitor of metalloproteinases (TIMPs) in heart failure. Heart Fail Rev. 2012, 17, 693–706.
- Gyöngyösi, M.; Winkler, J.; Ramos, I. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191.
- Ravassa, S.; Lopez, B.; Querejeta, R.; Echegaray, K.; San-Jose, G.; Moreno, M.U.; Beaumont, F.J.; Gonzalez, A.; Diez, J. Phenotyping of myocardial fibrosis in hypertensive patients with heart failure. Influence on clinical outcome. J. Hypertens. 2017, 35, 853–861.
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82.
- Bing, R.; Dweck, M.R. Myocardial fibrosis: Why image, how to image and clinical implications. Heart 2019, 105, 1832–1840.
- Lopez, B.; Ravassa, S.; Moreno, M.U.; San José, G.; Beaumont, J.; González, A.; Díez, J. Diffuse myocardial fibrosis: Mechanisms, diagnosis and therapeutic approaches. Nat. Rev. Cardiol. 2021, 18, 479–498.
- Espeland, T.; Lunde, I.G.; Amundsen, B.H.; Gullestad, L.; Aakhus, S. Myocardial fibrosis. Tidsskriftet 2018, 138.
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac fibrosis: The fibroblast awakens. Circ. Res. 2016, 118, 1021–1040.
- Liu, T.; Song, D.; Dong, J.; Zhu, P.; Liu, J.; Liu, W.; Ma, X.; Zhao, L.; Ling, S. Current unserstandings of the pathophysiology of myocardial fibrosis and its quantitative assessment in heart failure. Front. Physiol. 2017, 8, 238.
More
Information
Subjects:
Cardiac & Cardiovascular Systems; Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
965
Revisions:
2 times
(View History)
Update Date:
07 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No